
Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize


Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of the SAT Gene Family
2.2. Annotation of the SAT Genes
2.3. Transcriptome Analysis
2.4. Phylogenetic Tree Analyses
2.5. Data Source
3. Results
3.1. Identification of SAT Proteins in Maize B73 Inbred Line
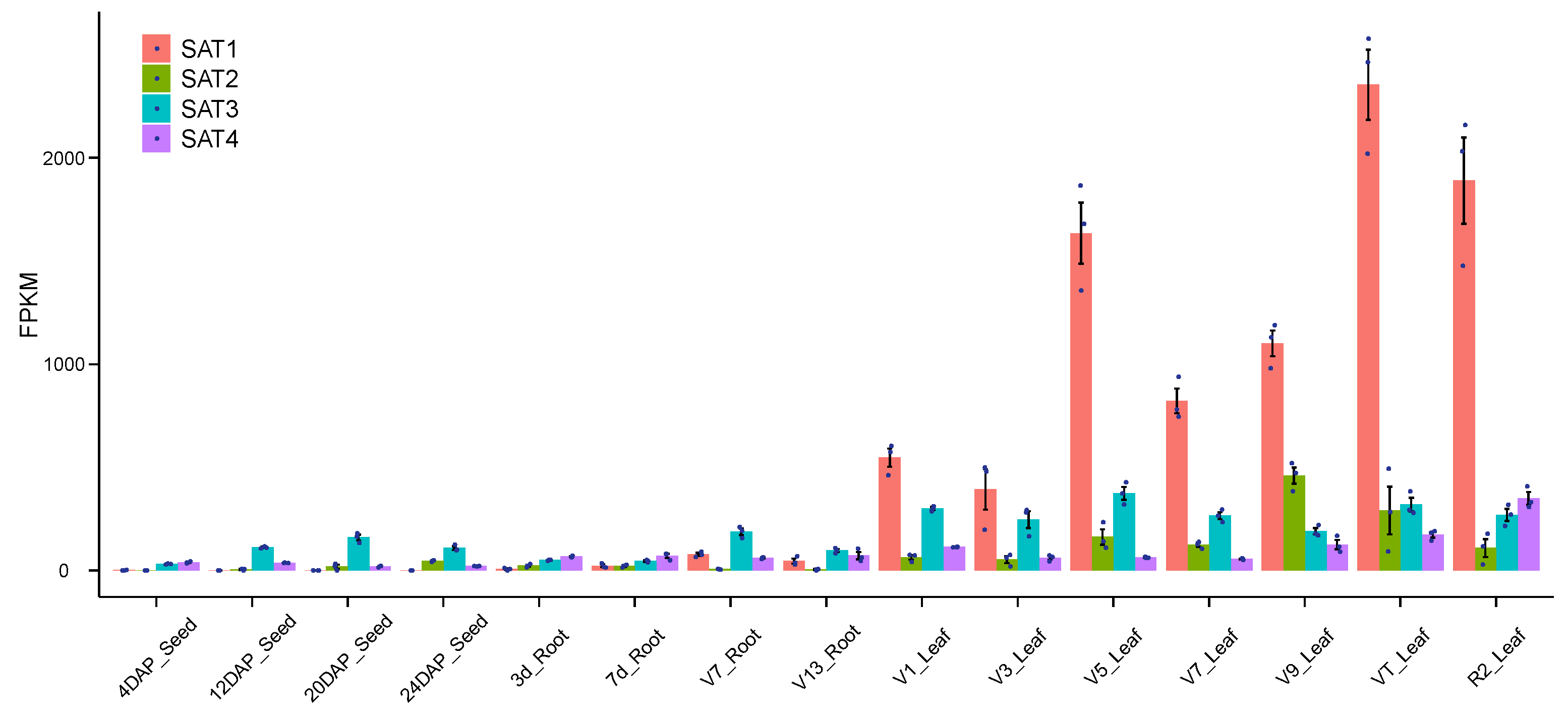
3.2. Expressional Profiling of the Maize SAT Genes
3.3. Structural Variation of SAT Genes in NAM Population
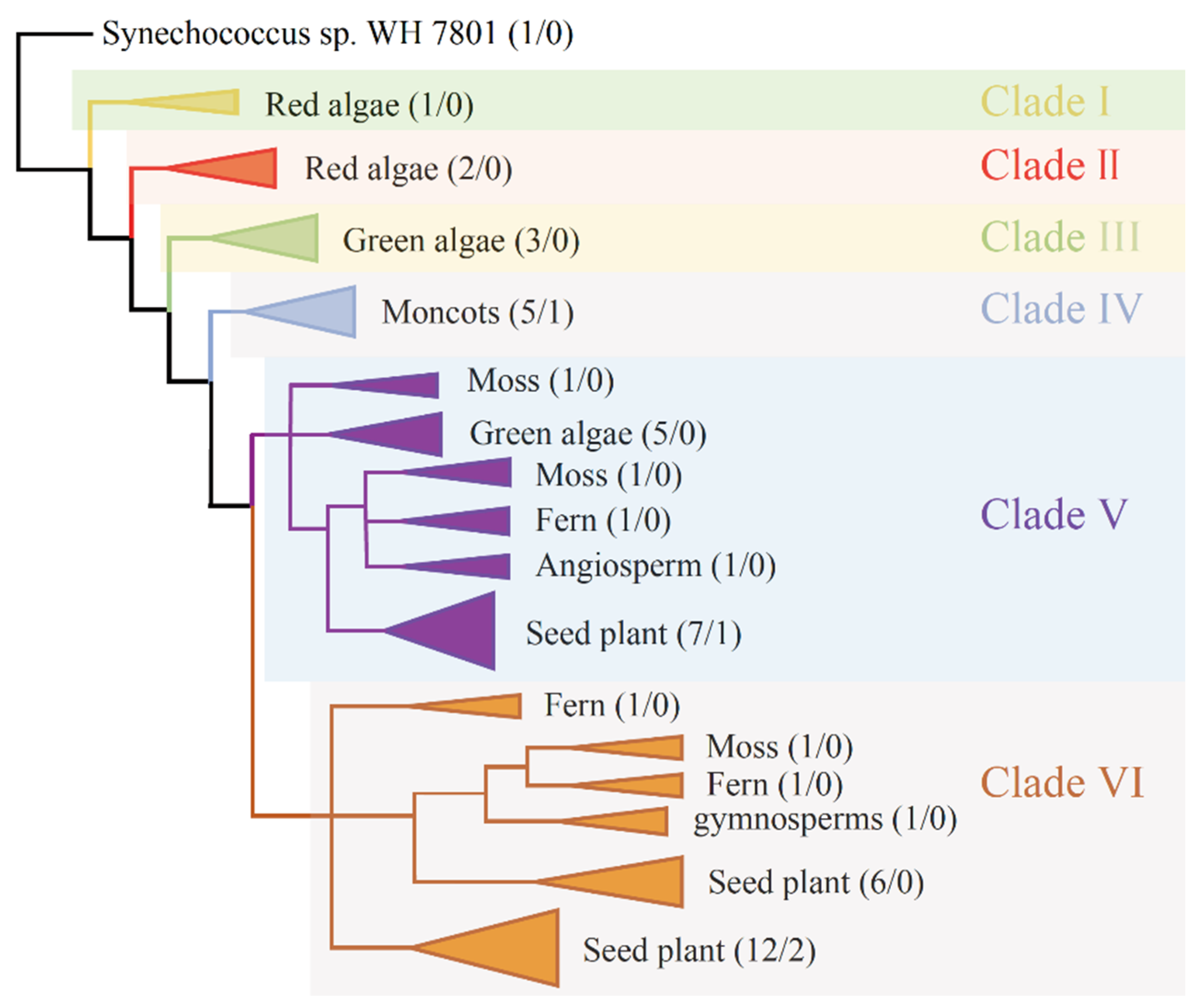
3.4. Evolutionary Analysis of SAT Genes
4. Discussion
4.1. Functional Redundancy of the SAT Gene Family
4.2. Transposable Elements and Gene Evolution
4.3. Structural Variation of the SAT Family in NAMs
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Messing, J. Proteome balancing of the maize seed for higher nutritional value. Front. Plant. Sci. 2014, 5, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, W.; Messing, J. Balancing of sulfur storage in maize seed. BMC Plant Biol. 2012, 12, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leustek, T.; Martin, M.N.; Bick, J.A.; Davies, J.P. Pathways and Regulation of Sulfur Metabolism Revealed through Molecular and Genetic Studies. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 141–165. [Google Scholar] [CrossRef]
- Kawashima, C.G.; Berkowitz, O.; Hell, R.; Noji, M.; Saito, K. Characterization and expression analysis of a serine acetyltransferase gene family involved in a key step of the sulfur assimilation pathway in Arabidopsis. Plant Physiol. 2005, 137, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Mochida, K.; Kato, T.; Tabata, S.; Yoshimoto, N.; Noji, M.; Saito, K. Comparative Genomics and Reverse Genetics Analysis Reveal Indispensable Functions of the Serine Acetyltransferase Gene Family in Arabidopsis. Plant Cell 2008, 20, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Planta, J.; Xiang, X.; Leustek, T.; Messing, J. Engineering sulfur storage in maize seed proteins without apparent yield loss. Proc. Natl. Acad. Sci. USA 2017, 114, 11386–11391. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Wu, Y.; Planta, J.; Messing, J.; Leustek, T. Overexpression of serine acetyltransferase in maize leaves increases seed-specific methionine-rich zeins. Plant. Biotechnol. J. 2018, 16, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Olsen, M.; Krone, T.; Phillips, R. BSSS53 as a Donor Source for Increased Whole-Kernel Methionine in Maize. Crop Sci. 2005, 43, 1634–1642. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.; Zhao, H.; Zhao, H.; Song, W.; Zhang, M.; Cui, Y.; Dong, X.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xiang, X.; Huang, Y.; Zhou, Y.; An, D.; Dong, J.; Zhao, C.; Liu, H.; Li, Y.; Wang, Q.; et al. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat. Commun. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef]
- McMullen, M.D.; Kresovich, S.; Villeda, H.S.; Bradbury, P.; Li, H.; Sun, Q.; Flint-Garcia, S.; Thornsberry, J.; Acharya, C.; Bottoms, C.; et al. Genetic properties of the maize nested association mapping population. Science 2009, 325, 737–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y.; et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stelpflug, S.C.; Sekhon, R.S.; Vaillancourt, B.; Hirsch, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. An Expanded Maize Gene Expression Atlas based on RNA Sequencing and its Use to Explore Root Development. Plant Genome 2016, 9. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Miao, Z.; Ren, C.; Yuan, R.; Tang, Y.; Zhang, X.; Han, Z.; Ma, C. Evolution of intron-poor clades and expression patterns of the glycosyltransferase family 47. Planta 2018, 247, 745–760. [Google Scholar] [CrossRef]
- Yeon, J.Y.; Yoo, S.J.; Takagi, H.; Kang, H.A. A Novel Mitochondrial Serine O-Acetyltransferase, OpSAT1, Plays a Critical Role in Sulfur Metabolism in the Thermotolerant Methylotrophic Yeast Ogataea parapolymorpha. Sci. Rep. 2018, 8, 2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howarth, J.R.; Dominguez-Solis, J.R.; Gutierrez-Alcala, G.; Wray, J.L.; Romero, L.C.; Gotor, C. The serine acetyltransferase gene family in Arabidopsis thaliana and the regulation of its expression by cadmium. Plant Mol. Biol. 2003, 51, 589–598. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, L.; Dai, J.; Li, J.; Yan, J. Identification and characterization of CACTA transposable elements capturing gene fragments in maize. Chin. Sci. Bull. 2009, 54, 642–651. [Google Scholar] [CrossRef]
- Liu, Z.; Li, X.; Wang, T.; Messing, J.; Xu, J.H. The Wukong Terminal-Repeat Retrotransposon in Miniature (TRIM) Elements in Diverse Maize Germplasm. G3 (Bethesda) 2015, 5, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Tavares, S.; Wirtz, M.; Beier, M.P.; Bogs, J.; Hell, R.; Amancio, S. Characterization of the serine acetyltransferase gene family of Vitis vinifera uncovers differences in regulation of OAS synthesis in woody plants. Front. Plant Sci. 2015, 6, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noji, M.; Takagi, Y.; Kimura, N.; Inoue, K.; Saito, M.; Horikoshi, M.; Saito, F.; Takahashi, H.; Saito, K. Serine acetyltransferase involved in cysteine biosynthesis from spinach: Molecular cloning, characterization and expression analysis of cDNA encoding a plastidic isoform. Plant Cell Physiol. 2001, 42, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, T.V.; Harrigan, G.G.; Perez, T.; Flint-Garcia, S. Compositional assessments of key maize populations: B73 hybrids of the Nested Association Mapping founder lines and diverse landrace inbred lines. J. Agric. Food Chem. 2015, 63, 5282–5295. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 2005, 15, 621–627. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fernie, A.R.; Yan, J. The Past, Present, and Future of Maize Improvement: Domestication, Genomics, and Functional Genomic Routes toward Crop Enhancement. Plant Commun. 2020, 1, 100010. [Google Scholar] [CrossRef] [PubMed]
- Cone, K.C.; Burr, F.A.; Burr, B. Molecular analysis of the maize anthocyanin regulatory locus C1. Proc. Natl. Acad. Sci. USA 1986, 83, 9631–9635. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, X.; Lin, Z.; Wang, J.; Liu, H.; Zhou, L.; Zhong, S.; Li, Y.; Zhu, C.; Lai, J.; et al. A Large Transposon Insertion in the stiff1 Promoter Increases Stalk Strength in Maize. Plant Cell 2020, 32, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Varagona, M.J.; Purugganan, M.; Wessler, S.R. Alternative splicing induced by insertion of retrotransposons into the maize waxy gene. Plant Cell 1992, 4, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Li, Z.; Li, W.; Ku, L.; Wang, C.; Ye, J.; Li, K.; Yang, N.; Li, Y.; Zhong, T.; et al. CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc. Natl. Acad. Sci. USA 2013, 110, 16969–16974. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Seetharam, A.S.; Chougule, K.; Ou, S.; Swentowsky, K.W.; Gent, J.I.; Llaca, V.; Woodhouse, M.; Manchanda, N.; Presting, G.G.; et al. Gapless assembly of maize chromosomes using long read technologies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [Green Version]
- Flint-Garcia, S.A.; Thuillet, A.C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Flint-Garcia, S.A.; Bodnar, A.L.; Scott, M.P. Wide variability in kernel composition, seed characteristics, and zein profiles among diverse maize inbreds, landraces, and teosinte. Theor. Appl. Genet. 2009, 119, 1129–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, T.V.; Chassy, A.W.; Fiehn, O.; Flint-Garcia, S.; Zeng, Q.; Skogerson, K.; Harrigan, G.G. Metabolomic Assessment of Key Maize Resources: GC-MS and NMR Profiling of Grain from B73 Hybrids of the Nested Association Mapping (NAM) Founders and of Geographically Diverse Landraces. J. Agric. Food Chem. 2016, 64, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tanskanen, J.; Immonen, S.; Nevo, E.; Schulman, A.H. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. USA 2000, 97, 6603–6607. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Lu, G.; Zhao, Q.; Liu, X.; Han, B. Genome-wide analysis of transposon insertion polymorphisms reveals intraspecific variation in cultivated rice. Plant Physiol. 2008, 148, 25–40. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Genbank ID | Gene Locus | Gene Size (bp) | Exon Number | CDS Length (bp) | Protein (aa) | Molecular Weight (kDa) |
|---|---|---|---|---|---|---|---|
| SAT1 | Zm00001d011735 | chr8:159516273-159517206 | 933 | 1 | 933 | 310 | 32.5 |
| SAT2 | Zm00001d028154 | chr1:24198740-24199685 | 945 | 1 | 945 | 314 | 32.4 |
| SAT3 | Zm00001d027536 | chr1:7652686-7639117 | 13,596 | 9 | 1023 | 340 | 36.4 |
| SAT4 | Zm00001d038737 | chr6:170865665-170873061 | 7396 | 3 | 1056 | 351 | 37.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Li, S.; Ji, C.; Zhou, Y.; Li, C.; Wang, W. Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize. Genes 2021, 12, 437. https://doi.org/10.3390/genes12030437
Zhao Z, Li S, Ji C, Zhou Y, Li C, Wang W. Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize. Genes. 2021; 12(3):437. https://doi.org/10.3390/genes12030437
Chicago/Turabian StyleZhao, Zhixuan, Shuai Li, Chen Ji, Yong Zhou, Changsheng Li, and Wenqin Wang. 2021. "Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize" Genes 12, no. 3: 437. https://doi.org/10.3390/genes12030437
APA StyleZhao, Z., Li, S., Ji, C., Zhou, Y., Li, C., & Wang, W. (2021). Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize. Genes, 12(3), 437. https://doi.org/10.3390/genes12030437