
Ribosomal RNA Transcription Regulation in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
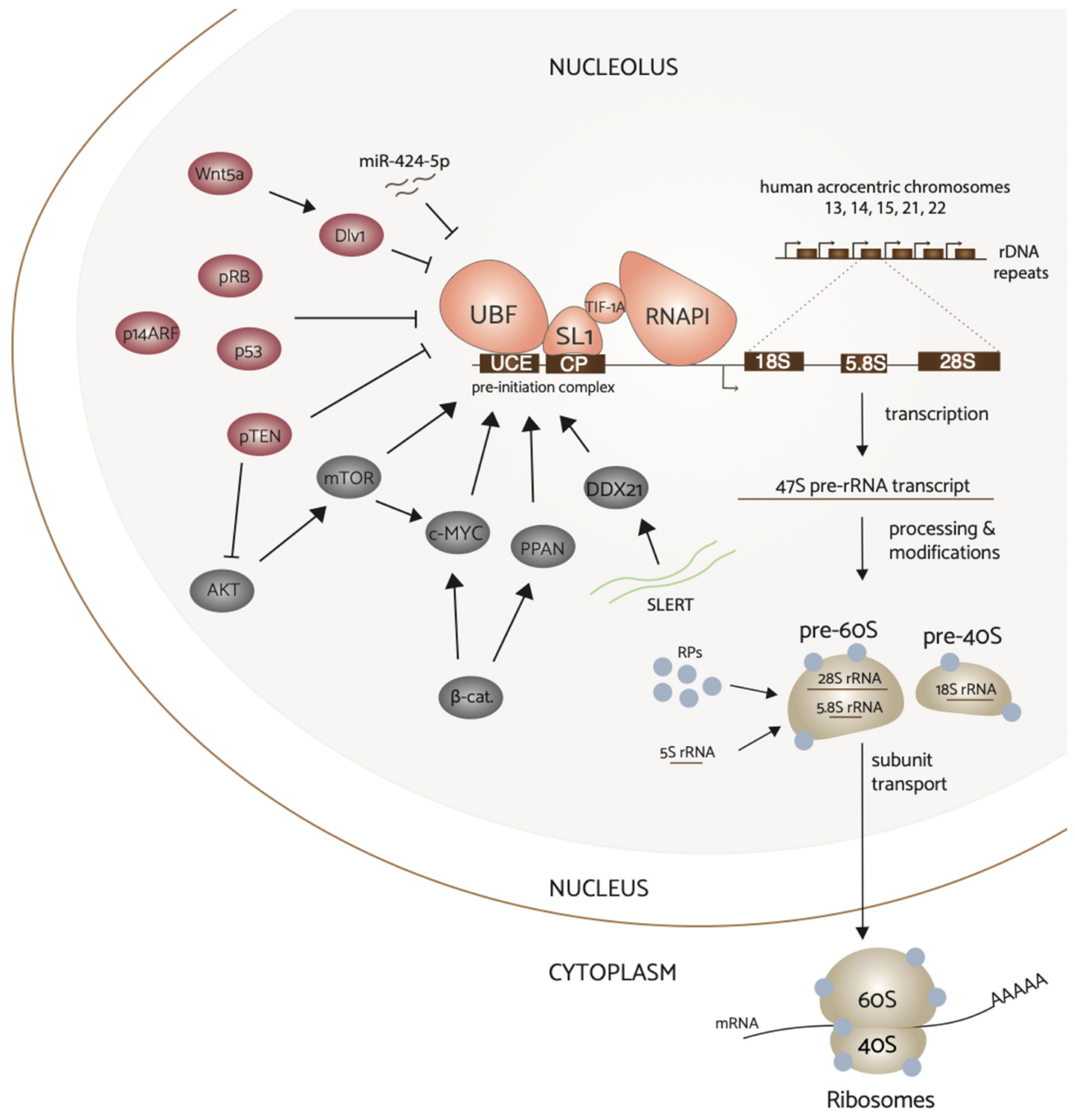
2. Ribosome Biogenesis
2.1. Ribosome Biogenesis
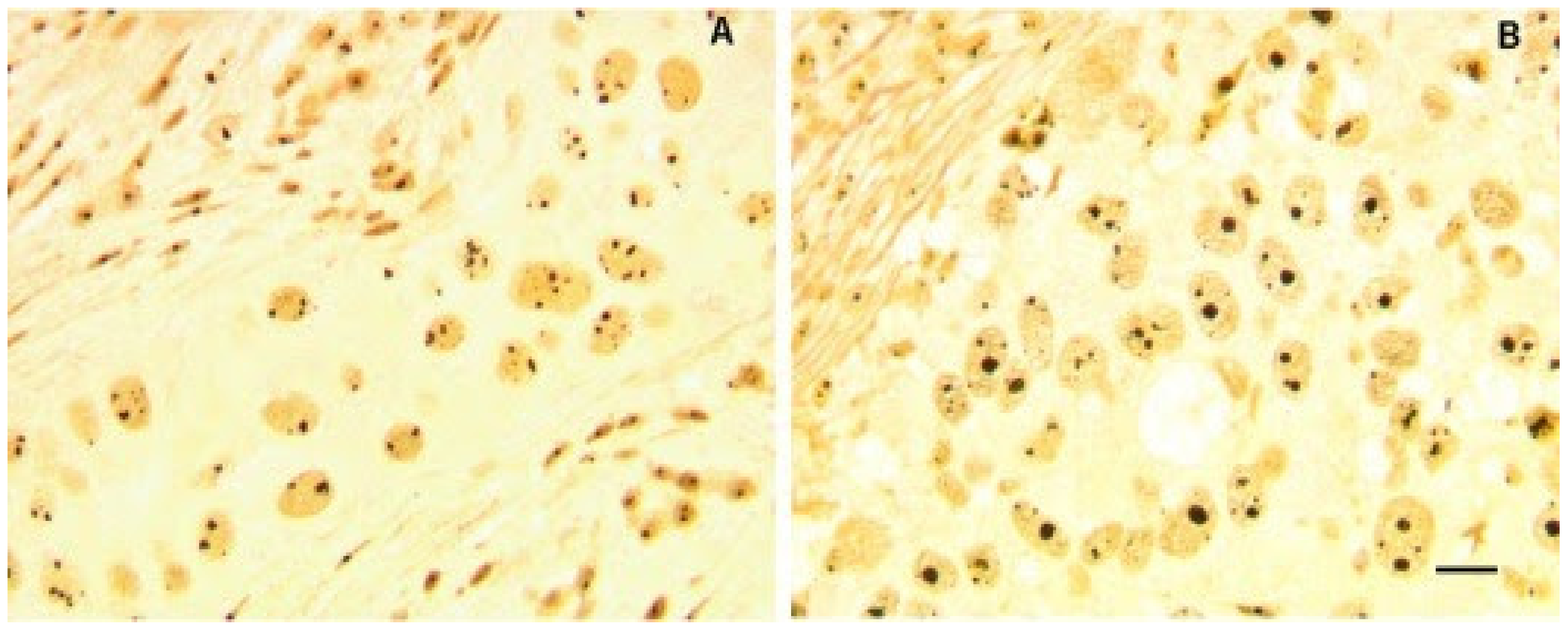
2.2. Nucleolar Remodeling in Breast Cancer
3. Signal Transduction Pathways Modulate RNAPI Activity
3.1. MYC
3.2. PI3K/AKT/mTOR
3.3. Wnt
4. Tumor Suppressor Proteins Inhibit rDNA Transcription
4.1. p53 & pRb
4.2. PTEN
4.3. ARF
5. Regulation of rRNA Synthesis by Noncoding RNAs
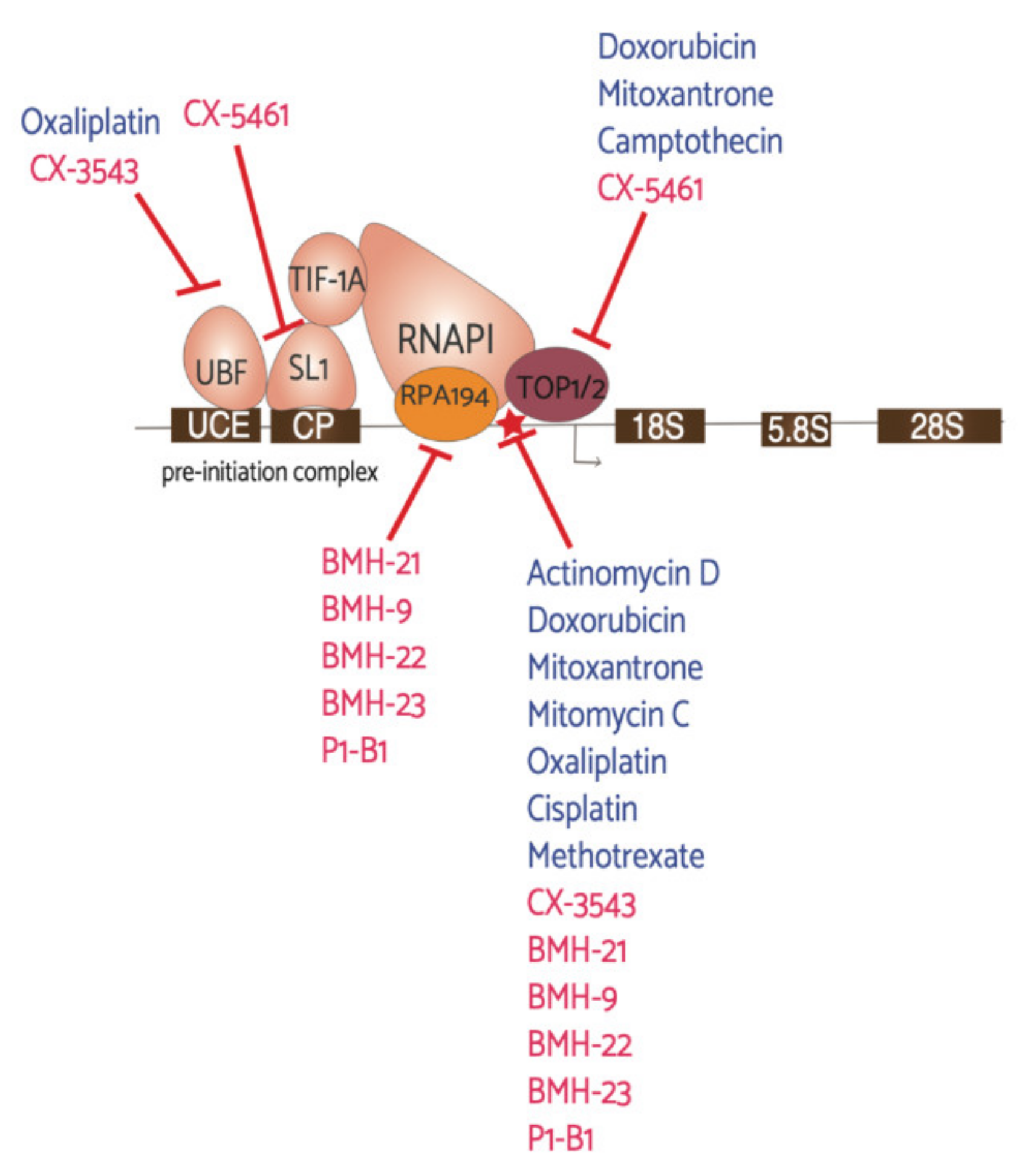
6. Therapeutic Targeting of rRNA Synthesis to Treat Breast Cancer
6.1. Current Anti-Cancer Drugs with Effects on RNAPI Activity
6.2. CX-3543
6.3. CX-5461
6.4. BMH-21 and Other BMH Molecules
6.5. Combination Drugs: P1-B1
7. Closing Remarks/Future Directions
Author Contributions
Funding
International Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [Green Version]
- Januskeviciene, I.; Petrikaite, V. Heterogeneity of breast cancer: The importance of interaction between different tumor cell populations. Life Sci. 2019, 239, 117009. [Google Scholar] [CrossRef]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Pianese, G. Beitraege zur Histologie und Aetiologie der Carconoms [Contributions to the histology and etiology of carcinomas]. Beitr. Pathol. Anat. Allgem. Pathol. 1896, 142, 1–193. [Google Scholar]
- Derenzini, M.; Montanaro, L.; Trere, D. Ribosome biogenesis and cancer. Acta Histochem. 2017, 119, 190–197. [Google Scholar] [CrossRef]
- Derenzini, M.; Betts, C.M.; Trere, D.; Mambelli, V.; Millis, R.R.; Eusebi, V.; Cancellieri, A. Diagnostic value of silver-stained interphasic nucleolar organizer regions in breast tumors. Ultrastruct. Pathol. 1990, 14, 233–245. [Google Scholar] [CrossRef]
- Elsharawy, K.A.; Toss, M.S.; Raafat, S.; Ball, G.; Green, A.R.; Aleskandarany, M.A.; Dalton, L.W.; Rakha, E.A. Prognostic significance of nucleolar assessment in invasive breast cancer. Histopathology 2020, 76, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Bustelo, X.R.; Dosil, M. Ribosome biogenesis and cancer: Basic and translational challenges. Curr. Opin. Genet. Dev. 2018, 48, 22–29. [Google Scholar] [CrossRef]
- Gaviraghi, M.; Vivori, C.; Tonon, G. How Cancer Exploits Ribosomal RNA Biogenesis: A Journey beyond the Boundaries of rRNA Transcription. Cells 2019, 8, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belin, S.; Beghin, A.; Solano-Gonzalez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.C.; Mertani, H.C.; Dumontet, C.; Diaz, J.J. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [Green Version]
- Henderson, A.S.; Warburton, D.; Atwood, K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA 1972, 69, 3394–3398. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Verdun, D. The nucleolus today. J. Cell Sci. 1991, 99(Pt. 3), 465–471. [Google Scholar] [PubMed]
- Thiry, M.; Cheutin, T.; O’Donohue, M.F.; Kaplan, H.; Ploton, D. Dynamics and three-dimensional localization of ribosomal RNA within the nucleolus. RNA 2000, 6, 1750–1761. [Google Scholar] [CrossRef] [Green Version]
- Moss, T.; Stefanovsky, V.Y. At the center of eukaryotic life. Cell 2002, 109, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Conconi, A.; Widmer, R.M.; Koller, T.; Sogo, J.M. Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 1989, 57, 753–761. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [Green Version]
- Valori, V.; Tus, K.; Laukaitis, C.; Harris, D.T.; LeBeau, L.; Maggert, K.A. Human rDNA copy number is unstable in metastatic breast cancers. Epigenetics 2020, 15, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Jantzen, H.M.; Admon, A.; Bell, S.P.; Tjian, R. Nucleolar transcription factor hUBF contains a DNA-binding motif with homology to HMG proteins. Nature 1990, 344, 830–836. [Google Scholar] [CrossRef]
- Bell, S.P.; Learned, R.M.; Jantzen, H.M.; Tjian, R. Functional cooperativity between transcription factors UBF1 and SL1 mediates human ribosomal RNA synthesis. Science 1988, 241, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Panov, K.I.; Friedrich, J.K.; Trinkle-Mulcahy, L.; Lamond, A.I.; Zomerdijk, J.C. hRRN3 is essential in the SL1-mediated recruitment of RNA Polymerase I to rRNA gene promoters. EMBO J. 2001, 20, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of RNA polymerase I transcription during disease. Biochim. Biophys. Acta 2013, 1829, 342–360. [Google Scholar] [CrossRef] [Green Version]
- Farley-Barnes, K.I.; McCann, K.L.; Ogawa, L.M.; Merkel, J.; Surovtseva, Y.V.; Baserga, S.J. Diverse Regulators of Human Ribosome Biogenesis Discovered by Changes in Nucleolar Number. Cell Rep. 2018, 22, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, L.M.; Buhagiar, A.F.; Abriola, L.; Leland, B.A.; Surovtseva, Y.V.; Baserga, S.J. Increased numbers of nucleoli in a genome-wide RNAi screen reveal proteins that link the cell cycle to RNA polymerase I transcription. Mol. Biol. Cell 2021. [Google Scholar] [CrossRef]
- MacCarty, W.C. Identification of the cancer cell. JAMA 1936, 107, 844–845. [Google Scholar] [CrossRef]
- Ploton, D.; Menager, M.; Jeannesson, P.; Himber, G.; Pigeon, F.; Adnet, J.J. Improvement in the staining and in the visualization of the argyrophilic proteins of the nucleolar organizer region at the optical level. Histochem. J. 1986, 18, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Goodpasture, C.; Bloom, S.E. Visualization of nucleolar organizer regions im mammalian chromosomes using silver staining. Chromosoma 1975, 53, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Pession, A.; Trere, D. Quantity of nucleolar silver-stained proteins is related to proliferating activity in cancer cells. Lab. Invest. 1990, 63, 137–140. [Google Scholar] [PubMed]
- Derenzini, M.; Ploton, D. Interphase nucleolar organizer regions in cancer cells. Int. Rev. Exp. Pathol. 1991, 32, 149–192. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Trere, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; White, R.J. MYC regulation of cell growth through control of transcription by RNA polymerases I and III. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Pattengale, P.K.; Leder, P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 1984, 38, 627–637. [Google Scholar] [CrossRef]
- Schoenenberger, C.A.; Andres, A.C.; Groner, B.; van der Valk, M.; LeMeur, M.; Gerlinger, P. Targeted c-myc gene expression in mammary glands of transgenic mice induces mammary tumours with constitutive milk protein gene transcription. EMBO J. 1988, 7, 169–175. [Google Scholar] [CrossRef]
- Qu, J.; Zhao, X.; Wang, J.; Liu, X.; Yan, Y.; Liu, L.; Cai, H.; Qu, H.; Lu, N.; Sun, Y.; et al. MYC overexpression with its prognostic and clinicopathological significance in breast cancer. Oncotarget 2017, 8, 93998–94008. [Google Scholar] [CrossRef]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [Green Version]
- Poortinga, G.; Wall, M.; Sanij, E.; Siwicki, K.; Ellul, J.; Brown, D.; Holloway, T.P.; Hannan, R.D.; McArthur, G.A. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res. 2011, 39, 3267–3281. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q.; et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef]
- Potapova, T.A.; Unruh, J.R.; Yu, Z.; Rancati, G.; Li, H.; Stampfer, M.R.; Gerton, J.L. Superresolution microscopy reveals linkages between ribosomal DNA on heterologous chromosomes. J. Cell Biol. 2019, 218, 2492–2513. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, J.; Park, B.H.; Wolff, A.C. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J. Natl. Compr. Cancer Netw. 2013, 11, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.K.; Liu, H.; Zheng, X.F. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle 2010, 9, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Nguyen le, X.T.; Mitchell, B.S. Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc. Natl. Acad. Sci. USA 2013, 110, 20681–20686. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, S.; Wierzbicki, A.J.; Sacchi, N. Mammary epithelial morphogenesis and early breast cancer. Evidence of involvement of basal components of the RNA Polymerase I transcription machinery. Cell Cycle 2016, 15, 2515–2526. [Google Scholar] [CrossRef] [Green Version]
- Van Schie, E.H.; van Amerongen, R. Aberrant WNT/CTNNB1 Signaling as a Therapeutic Target in Human Breast Cancer: Weighing the Evidence. Front. Cell Dev. Biol. 2020, 8, 25. [Google Scholar] [CrossRef]
- Pfister, A.S.; Kuhl, M. Of Wnts and Ribosomes. Prog Mol. Biol. Transl. Sci. 2018, 153, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Huo, D.; Khramtsov, A.; Khramtsova, G.; Zhang, C.; Goss, K.H.; Olopade, O.I. Beta-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol. Carcinog. 2016, 55, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.E.; Kammerud, S.C.; Metge, B.J.; AlSheikh, H.A.; Schneider, D.A.; Chen, D.; Samant, R.S. Inhibiting beta-catenin disables nucleolar functions in triple-negative breast cancer. Cell Death Dis. 2021, 12, 242. [Google Scholar] [CrossRef]
- Bao, L.; Guo, T.; Wang, J.; Zhang, K.; Bao, M. Prognostic genes of triple-negative breast cancer identified by weighted gene co-expression network analysis. Oncol. Lett. 2020, 19, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, A.S.; Keil, M.; Kuhl, M. The Wnt Target Protein Peter Pan Defines a Novel p53-independent Nucleolar Stress-Response Pathway. J. Biol. Chem. 2015, 290, 10905–10918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannheisig, D.P.; Bachle, J.; Tasic, J.; Keil, M.; Pfister, A.S. The Wnt/beta-Catenin Pathway is Activated as a Novel Nucleolar Stress Response. J. Mol. Biol. 2021, 433, 166719. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.P.; Manchanda, M.; Mohapatra, P.; Andersson, T. WNT5A as a therapeutic target in breast cancer. Cancer Metastasis Rev. 2018, 37, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dass, R.A.; Sarshad, A.A.; Carson, B.B.; Feenstra, J.M.; Kaur, A.; Obrdlik, A.; Parks, M.M.; Prakash, V.; Love, D.K.; Pietras, K.; et al. Wnt5a Signals through DVL1 to Repress Ribosomal DNA Transcription by RNA Polymerase I. PLoS Genet. 2016, 12, e1006217. [Google Scholar] [CrossRef]
- Chen, S.; Seiler, J.; Santiago-Reichelt, M.; Felbel, K.; Grummt, I.; Voit, R. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol. Cell 2013, 52, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Mukherjee, M.; Johnson, J.P.; Patel, M.; Huey, B.; Albertson, D.G.; Simin, K. Cooperativity of Rb, Brca1, and p53 in malignant breast cancer evolution. PLoS Genet. 2012, 8, e1003027. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef] [Green Version]
- Budde, A.; Grummt, I. p53 represses ribosomal gene transcription. Oncogene 1999, 18, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Voit, R.; Schafer, K.; Grummt, I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol. Cell Biol. 1997, 17, 4230–4237. [Google Scholar] [CrossRef] [Green Version]
- Hannan, K.M.; Hannan, R.D.; Smith, S.D.; Jefferson, L.S.; Lun, M.; Rothblum, L.I. Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1. Oncogene 2000, 19, 4988–4999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanaugh, A.H.; Hempel, W.M.; Taylor, L.J.; Rogalsky, V.; Todorov, G.; Rothblum, L.I. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 1995, 374, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Trere, D.; Ceccarelli, C.; Montanaro, L.; Tosti, E.; Derenzini, M. Nucleolar size and activity are related to pRb and p53 status in human breast cancer. J. Histochem. Cytochem. 2004, 52, 1601–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derenzini, M.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Trere, D. The prognostic value of the AgNOR parameter in human breast cancer depends on the pRb and p53 status. J. Clin. Pathol. 2004, 57, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.S.; Zeng, S.X.; Jin, Y.; Sun, X.X.; David, L.; Lu, H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell Biol. 2004, 24, 7654–7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [Green Version]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008, 112, 5228–5237. [Google Scholar] [CrossRef] [Green Version]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [Green Version]
- Lebok, P.; Kopperschmidt, V.; Kluth, M.; Hube-Magg, C.; Ozden, C.; Taskin, B.; Hussein, K.; Mittenzwei, A.; Lebeau, A.; Witzel, I.; et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer 2015, 15, 963. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Comai, L.; Johnson, D.L. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol. Cell Biol. 2005, 25, 6899–6911. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.P.; Smith, W.M.; Dahia, P.L.; Ziebold, U.; Gil, E.; Lees, J.A.; Eng, C. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999, 59, 5808–5814. [Google Scholar] [PubMed]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z.; Yang, J. Loss of PTEN expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef] [Green Version]
- Normand, G.; Hemmati, P.G.; Verdoodt, B.; von Haefen, C.; Wendt, J.; Guner, D.; May, E.; Dorken, B.; Daniel, P.T. p14ARF induces G2 cell cycle arrest in p53- and p21-deficient cells by down-regulating p34cdc2 kinase activity. J. Biol. Chem. 2005, 280, 7118–7130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayrault, O.; Andrique, L.; Fauvin, D.; Eymin, B.; Gazzeri, S.; Seite, P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene 2006, 25, 7577–7586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayrault, O.; Andrique, L.; Larsen, C.J.; Seite, P. Human Arf tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene 2004, 23, 8097–8104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, F.; Morin, F.; Ivanchuk, S.; Langlois, F.; Stefanovsky, V.; Rutka, J.; Moss, T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell 2010, 38, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Guibert, S.; Tiwari, V.K.; Ohlsson, R.; Langst, G. Epigenetic regulation of TTF-I-mediated promoter-terminator interactions of rRNA genes. EMBO J. 2008, 27, 1255–1265. [Google Scholar] [CrossRef]
- Pare, R.; Shin, J.S.; Lee, C.S. Increased expression of senescence markers p14(ARF) and p16(INK4a) in breast cancer is associated with an increased risk of disease recurrence and poor survival outcome. Histopathology 2016, 69, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Silva, J.M.; Dominguez, G.; Garcia, J.M.; Cantos, B.; Rodriguez, R.; Larrondo, F.J.; Provencio, M.; Espana, P.; Bonilla, F. Concomitant expression of p16INK4a and p14ARF in primary breast cancer and analysis of inactivation mechanisms. J. Pathol. 2003, 199, 289–297. [Google Scholar] [CrossRef] [PubMed]
- McCool, M.A.; Bryant, C.J.; Baserga, S.J. MicroRNAs and long non-coding RNAs as novel regulators of ribosome biogenesis. Biochem. Soc. Trans. 2020, 48, 595–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Lee, S.Y.; Gutierrez, A.; Perrigoue, J.; Thapa, R.J.; Tu, Z.; Jeffers, J.R.; Rhodes, M.; Anderson, S.; Oravecz, T.; et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood 2012, 120, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Challagundla, K.B.; Sun, X.X.; Zhang, X.; DeVine, T.; Zhang, Q.; Sears, R.C.; Dai, M.S. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell Biol. 2011, 31, 4007–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.M.; Zhou, X.; Gatignol, A.; Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 2014, 33, 4916–4923. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hao, Q.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal protein S14 negatively regulates c-Myc activity. J. Biol. Chem. 2013, 288, 21793–21801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Challagundla, K.B.; Sun, X.X.; Zhang, Q.; Dai, M.S. MicroRNA-130a associates with ribosomal protein L11 to suppress c-Myc expression in response to UV irradiation. Oncotarget 2015, 6, 1101–1114. [Google Scholar] [CrossRef]
- Connolly, M.; Paul, R.; Farre-Garros, R.; Natanek, S.A.; Bloch, S.; Lee, J.; Lorenzo, J.P.; Patel, H.; Cooper, C.; Sayer, A.A.; et al. miR-424-5p reduces ribosomal RNA and protein synthesis in muscle wasting. J. Cachexia Sarcopenia Muscle 2018, 9, 400–416. [Google Scholar] [CrossRef]
- Bublik, D.R.; Bursac, S.; Sheffer, M.; Orsolic, I.; Shalit, T.; Tarcic, O.; Kotler, E.; Mouhadeb, O.; Hoffman, Y.; Fuchs, G.; et al. Regulatory module involving FGF13, miR-504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E496–E505. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, J.; Wang, M.; Li, X.; Gong, H.; Tang, H.; Chen, L.; Wan, L.; Liu, Q. Activity dependent LoNA regulates translation by coordinating rRNA transcription and methylation. Nat. Commun. 2018, 9, 1726. [Google Scholar] [CrossRef]
- Bierhoff, H.; Schmitz, K.; Maass, F.; Ye, J.; Grummt, I. Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 357–364. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Dammert, M.A.; Grummt, I.; Bierhoff, H. lncRNA-Induced Nucleosome Repositioning Reinforces Transcriptional Repression of rRNA Genes upon Hypotonic Stress. Cell Rep. 2016, 14, 1876–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Senturk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.H.; Yao, R.W.; Zhang, Y.; Guo, C.J.; Jiang, S.; Xu, G.; Dong, R.; Yang, L.; Chen, L.L. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017, 169, 664–678.e616. [Google Scholar] [CrossRef] [PubMed]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Golomb, L.; Volarevic, S.; Oren, M. p53 and ribosome biogenesis stress: The essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Moore, H.M.; Enback, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hallstrom, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef] [PubMed]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef] [Green Version]
- Trask, D.K.; Muller, M.T. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc. Natl. Acad. Sci. USA 1988, 85, 1417–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kirdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef] [Green Version]
- Awad, D.; Prattes, M.; Kofler, L.; Rossler, I.; Loibl, M.; Pertl, M.; Zisser, G.; Wolinski, H.; Pertschy, B.; Bergler, H. Inhibiting eukaryotic ribosome biogenesis. BMC Biol. 2019, 17, 46. [Google Scholar] [CrossRef]
- Busche, M.; Scarpin, M.R.; Hnasko, R.; Brunkard, J.O. TOR coordinates nucleotide availability with ribosome biogenesis in plants. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Jordan, P.; Carmo-Fonseca, M. Cisplatin inhibits synthesis of ribosomal RNA in vivo. Nucleic Acids Res. 1998, 26, 2831–2836. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, N.; Herdman, C.; Mars, J.C.; Stefanovsky, V.; Tremblay, M.G.; Moss, T. Depletion of the cisplatin targeted HMGB-box factor UBF selectively induces p53-independent apoptotic death in transformed cells. Oncotarget 2015, 6, 27519–27536. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.X.; Xu, B.H.; Zhang, Y. CX-3543 Promotes Cell Apoptosis through Downregulation of CCAT1 in Colon Cancer Cells. Biomed. Res. Int. 2018, 2018, 9701957. [Google Scholar] [CrossRef]
- Papadopoulos, K.; Mita, A.; Ricart, A.; Hufnagel, D.; Northfelt, D.; Von Hoff, D.; Darjania, L.; Lim, J.; Padgett, C.; Marschke, R. Pharmacokinetic findings from the phase I study of Quarfloxin (CX-3543): A protein-rDNA quadruplex inhibitor, in patients with advanced solid tumors. Mol. Cancer Ther. 2007, 6, B93. [Google Scholar]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.S.; Brown, P. Transient rRNA synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 2015, 6, 34846–34858. [Google Scholar] [CrossRef] [Green Version]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Mars, J.C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Hilton, J.; Cescon, D.W.; Bedard, P.; Ritter, H.; Soong, J.; Gelmon, K.; Aparicio, S.; Seymour, L. CCTG IND.231: A phase 1 trial evaluating CX-5461 in patients with advanced solid tumors. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Zhang, Z.; Af Hallstrom, T.; Moore, H.M.; Sirajuddin, P.; Laiho, M. Small molecule BMH-compounds that inhibit RNA polymerase I and cause nucleolar stress. Mol. Cancer Ther. 2014, 13, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.L.; Zhao, C.L.; Chen, Q.; Xu, K.; Qiao, X.; Xu, J.Y. Targeting RNA polymerase I transcription machinery in cancer cells by a novel monofunctional platinum-based agent. Eur. J. Med. Chem. 2018, 155, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Scull, C.E.; Zhang, Y.; Tower, N.; Rasmussen, L.; Padmalayam, I.; Hunter, R.; Zhai, L.; Bostwick, R.; Schneider, D.A. Discovery of novel inhibitors of ribosome biogenesis by innovative high throughput screening strategies. Biochem. J. 2019, 476, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Awuah, S.G. A cell-based screening system for RNA polymerase I inhibitors. MedChemComm 2019, 10, 1765–1774. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harold, C.M.; Buhagiar, A.F.; Cheng, Y.; Baserga, S.J. Ribosomal RNA Transcription Regulation in Breast Cancer. Genes 2021, 12, 502. https://doi.org/10.3390/genes12040502
Harold CM, Buhagiar AF, Cheng Y, Baserga SJ. Ribosomal RNA Transcription Regulation in Breast Cancer. Genes. 2021; 12(4):502. https://doi.org/10.3390/genes12040502
Chicago/Turabian StyleHarold, Cecelia M., Amber F. Buhagiar, Yan Cheng, and Susan J. Baserga. 2021. "Ribosomal RNA Transcription Regulation in Breast Cancer" Genes 12, no. 4: 502. https://doi.org/10.3390/genes12040502
APA StyleHarold, C. M., Buhagiar, A. F., Cheng, Y., & Baserga, S. J. (2021). Ribosomal RNA Transcription Regulation in Breast Cancer. Genes, 12(4), 502. https://doi.org/10.3390/genes12040502