Loss-of-Function Variants in EFEMP1 Cause a Recognizable Connective Tissue Disorder Characterized by Cutis Laxa and Multiple Herniations

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Consent
2.2. Molecular Analysis
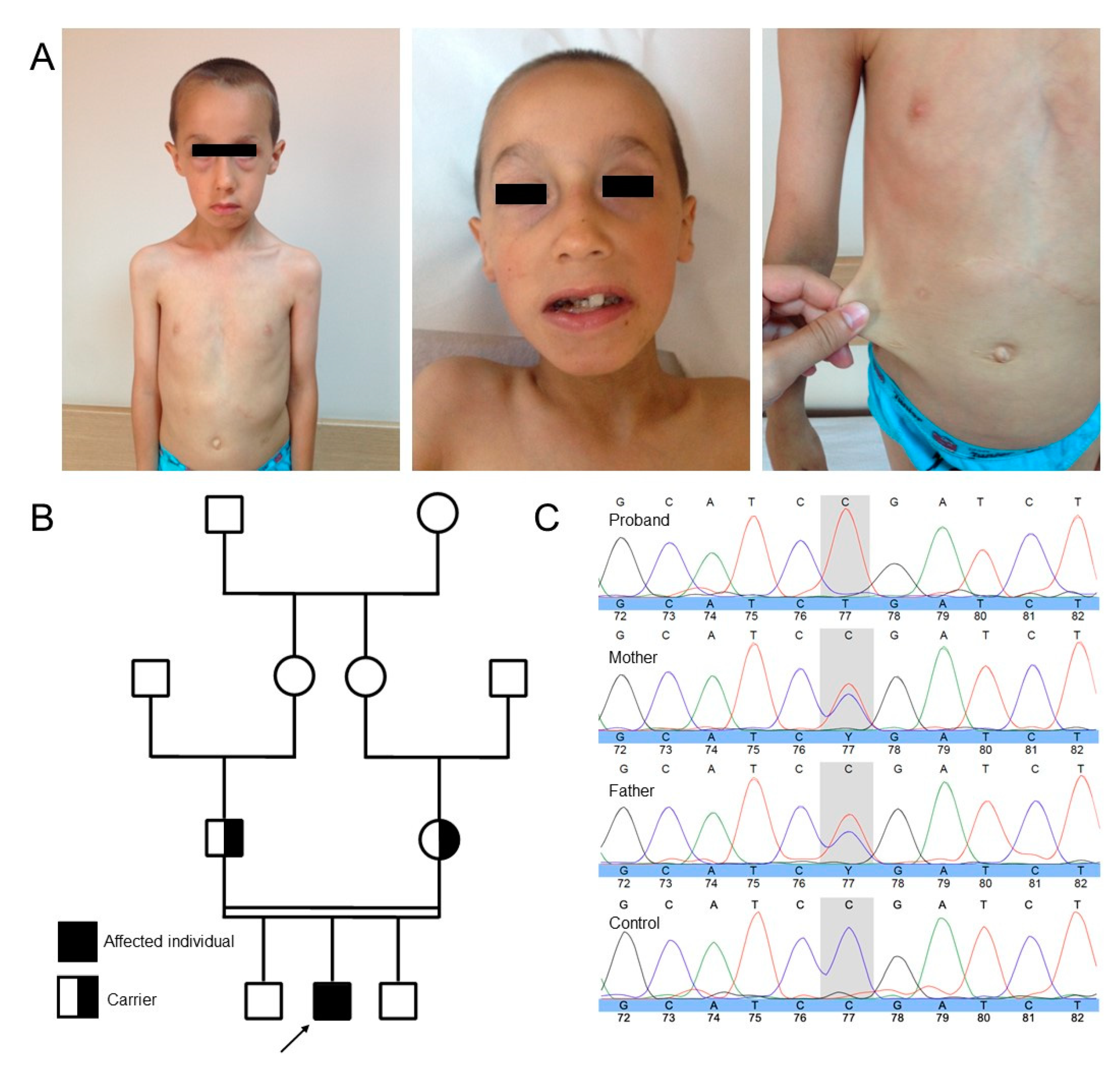
3. Case Presentation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy-Ryan, M.; Psychogios, A.; Lindor, N.M. Hereditary disorders of connective tissue: A guide to the emerging differential diagnosis. Genet. Med. 2010, 12, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Elahi, E.; Kalhor, R.; Banihosseini, S.S.; Torabi, N.; Pour-Jafari, H.; Houshmand, M.; Amini, S.S.; Ramezani, A.; Loeys, B. Homozygous missense mutation in fibulin-5 in an iranian autosomal recessive cutis laxa pedigree and associated haplotype. J. Investig. Dermatol. 2006, 126, 1506–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hucthagowder, V.; Sausgruber, N.; Kim, K.H.; Angle, B.; Marmorstein, L.Y.; Urban, Z. Fibulin-4: A novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 2006, 78, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, H.; Davis, E.C. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Biol. 2010, 42, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, I.; Uversky, V.N.; Furniss, D.; Wiberg, A. The Pathophysiological Significance of Fibulin-3. Biomolecules 2020, 10, 1294. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Lotery, A.J.; Munier, F.L.; Héon, E.; Piguet, B.; Guymer, R.H.; VanDenburgh, K.; Cousin, P.; Nishimura, D.; Swiderski, R.E.; et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999, 22, 199–202. [Google Scholar] [CrossRef]
- Bizzari, S.; El-Bazzal, L.; Nair, P.; Younan, A.; Stora, S.; Mehawej, C.; El-Hayek, S.; Delague, V.; Mégarbané, A. Recessive marfanoid syndrome with herniation associated with a homozygous mutation in Fibulin-3. Eur. J. Med. Genet. 2020, 63, 103869. [Google Scholar] [CrossRef]
- Driver, S.G.W.; Jackson, M.R.; Richter, K.; Tomlinson, P.; Brockway, B.; Halliday, B.J.; Markie, D.M.; Robertson, S.P.; Wade, E.M. Biallelic variants in EFEMP1 in a man with a pronounced connective tissue phenotype. Eur. J. Hum. Genet. 2019, 28, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoeldi, J.; Wang, Q.S.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; on behalf of the ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M.; et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Callewaert, B.; Su, C.-T.; Van Damme, T.; Vlummens, P.; Malfait, F.; Vanakker, O.; Schulz, B.; Mac Neal, M.; Davis, E.C.; Lee, J.G.; et al. Comprehensive Clinical and Molecular Analysis of 12 Families with Type 1 Recessive Cutis Laxa. Hum. Mutat. 2013, 34, 111–121. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Bakall, B.; Choi, J.; Liu, Z.; Sasaki, T.; Davis, E.C.; Marmorstein, A.D.; Marmorstein, L.Y. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum. Mol. Genet. 2007, 16, 3059–3070. [Google Scholar] [CrossRef]
- Megarbane, A.; Hanna, N.; Chouery, E.; Jalkh, N.; Mehawej, C.; Boileau, C. Marfanoid habitus, inguinal hernia, advanced bone age, and distinctive facial features: A new collagenopathy? Am. J. Med. Genet. Part A 2012, 158, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Lefeber, D.J.; Urban, Z.; De Meirleir, L.; Meinecke, P.; Kaesbach, G.G.; SykutCegielska, J.; Adamowicz, M.; Salafsky, I.; Ranells, J.D.; et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur. J. Hum. Genet. 2007, 16, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Kivuva, E.C.; Parker, M.J.; Cohen, M.C.; Wagner, B.E.; Sobey, G. De Barsy syndrome: A review of the phenotype. Clin. Dysmorphol. 2008, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Beyens, A.; Boel, A.; Symoens, S.; Callewaert, B. Cutis laxa: A comprehensive overview of clinical characteristics and pathophysiology. Clin. Genet. 2021, 99, 53–66. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| EFEMP1-Related CL | ARCL1a | ARCL1b | ARCL1c | ||||
|---|---|---|---|---|---|---|---|
| This study | Bizzari et al. [7] | Driver et al. [8] | Beyens et al. [24] | ||||
| Mégarbané et al. [21] | |||||||
| Clinical characteristics | |||||||
| Craniofacial dysmorphism | + a | + a | + a | + a | + b | + c | + d |
| Dental crowding | − | + | + | − | − | − | − |
| Cutis laxa | + | − | − | − | + | + | + |
| Thin translucent skin | + | − | − | + | − | − | + |
| Diaphragmatic hernia | + | − | + | + | + | + | + |
| Inguinal hernia | + | + | + | + | + | + | + |
| Hypermobile joints | + | + | + | + | + | + | + |
| Muscle hypotonia | + | + | + | + | − | − | + |
| Scoliosis | − | − | + | + | − | + | − |
| Pectus deformities | − | + | + | + | − | + | + |
| Tall Stature | − | + | + | + | − | − | − |
| Aortopathy | − | − | − | − | + | + | + |
| Emphysema | − | − | − | + | + | − | + |
| Gastrointestinal abnormalities | − | + | + | − | − | − | + |
| Bladder diverticula | − | + | + | + | + | − | + |
| Molecular characteristics | EFEMP1 | EFEMP1 | EFEMP1 | EFEMP1 | FBLN5 | FBLN4 | LTBP4 |
| cDNA level | c.1201C > T | c.163T > C | c.163T > C | c.320_324delTG GCA c. 615T > A | |||
| Protein level | p.(Arg401*) | p.(Cys55Arg) | p.(Cys55Arg) | p. (Met107fs) p.(Tyr205*) | |||
| Zygosity | Homozygous | Homozygous | Homozygous | Heterozygous | |||
| Other | VCPKMT and MYO3 variants | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verlee, M.; Beyens, A.; Gezdirici, A.; Gulec, E.Y.; Pottie, L.; De Feyter, S.; Vanhooydonck, M.; Tapaneeyaphan, P.; Symoens, S.; Callewaert, B. Loss-of-Function Variants in EFEMP1 Cause a Recognizable Connective Tissue Disorder Characterized by Cutis Laxa and Multiple Herniations. Genes 2021, 12, 510. https://doi.org/10.3390/genes12040510
Verlee M, Beyens A, Gezdirici A, Gulec EY, Pottie L, De Feyter S, Vanhooydonck M, Tapaneeyaphan P, Symoens S, Callewaert B. Loss-of-Function Variants in EFEMP1 Cause a Recognizable Connective Tissue Disorder Characterized by Cutis Laxa and Multiple Herniations. Genes. 2021; 12(4):510. https://doi.org/10.3390/genes12040510
Chicago/Turabian StyleVerlee, Maxim, Aude Beyens, Alper Gezdirici, Elif Yilmaz Gulec, Lore Pottie, Silke De Feyter, Michiel Vanhooydonck, Piyanoot Tapaneeyaphan, Sofie Symoens, and Bert Callewaert. 2021. "Loss-of-Function Variants in EFEMP1 Cause a Recognizable Connective Tissue Disorder Characterized by Cutis Laxa and Multiple Herniations" Genes 12, no. 4: 510. https://doi.org/10.3390/genes12040510
APA StyleVerlee, M., Beyens, A., Gezdirici, A., Gulec, E. Y., Pottie, L., De Feyter, S., Vanhooydonck, M., Tapaneeyaphan, P., Symoens, S., & Callewaert, B. (2021). Loss-of-Function Variants in EFEMP1 Cause a Recognizable Connective Tissue Disorder Characterized by Cutis Laxa and Multiple Herniations. Genes, 12(4), 510. https://doi.org/10.3390/genes12040510