
Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study System and Sampling
2.2. Genomic Data and Local Ancestry Inference
2.3. Describing Genome-Wide Variation of Domestic Ancestry and Genetic Diversity
2.4. Evaluating the Influence of Genetic Diversity, Stocking Intensity and Recombination Rate on Genomewide Variation in Domestic Ancestry
3. Results
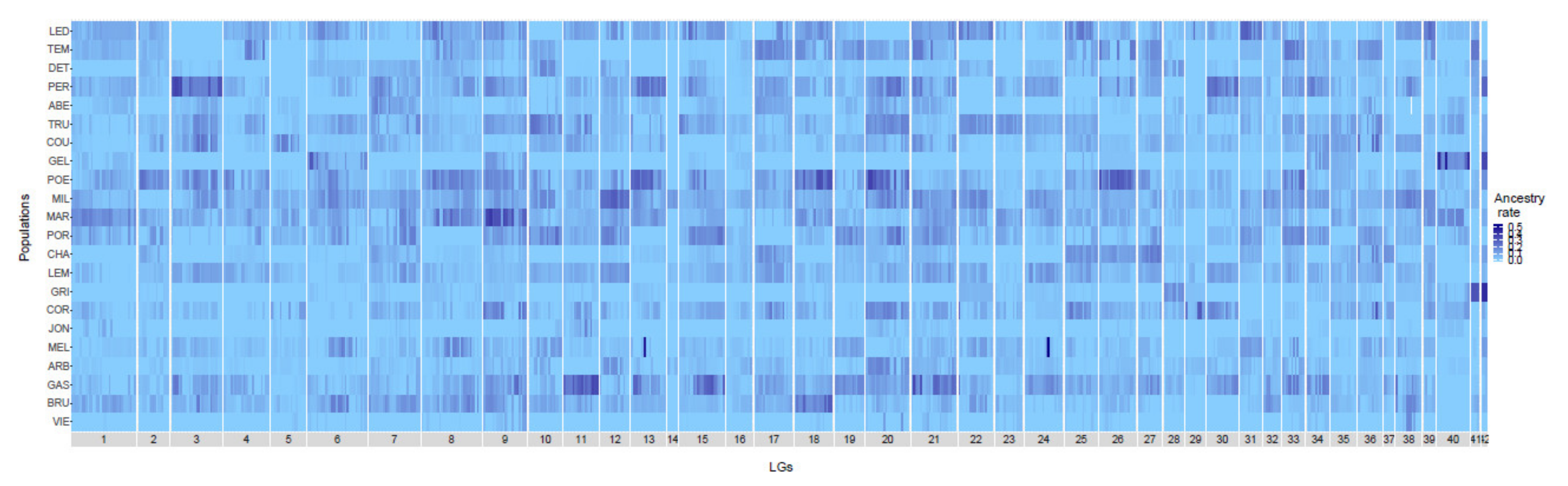
3.1. Genome-Wide Variation of Domestic Ancestry and Genetic Diversity
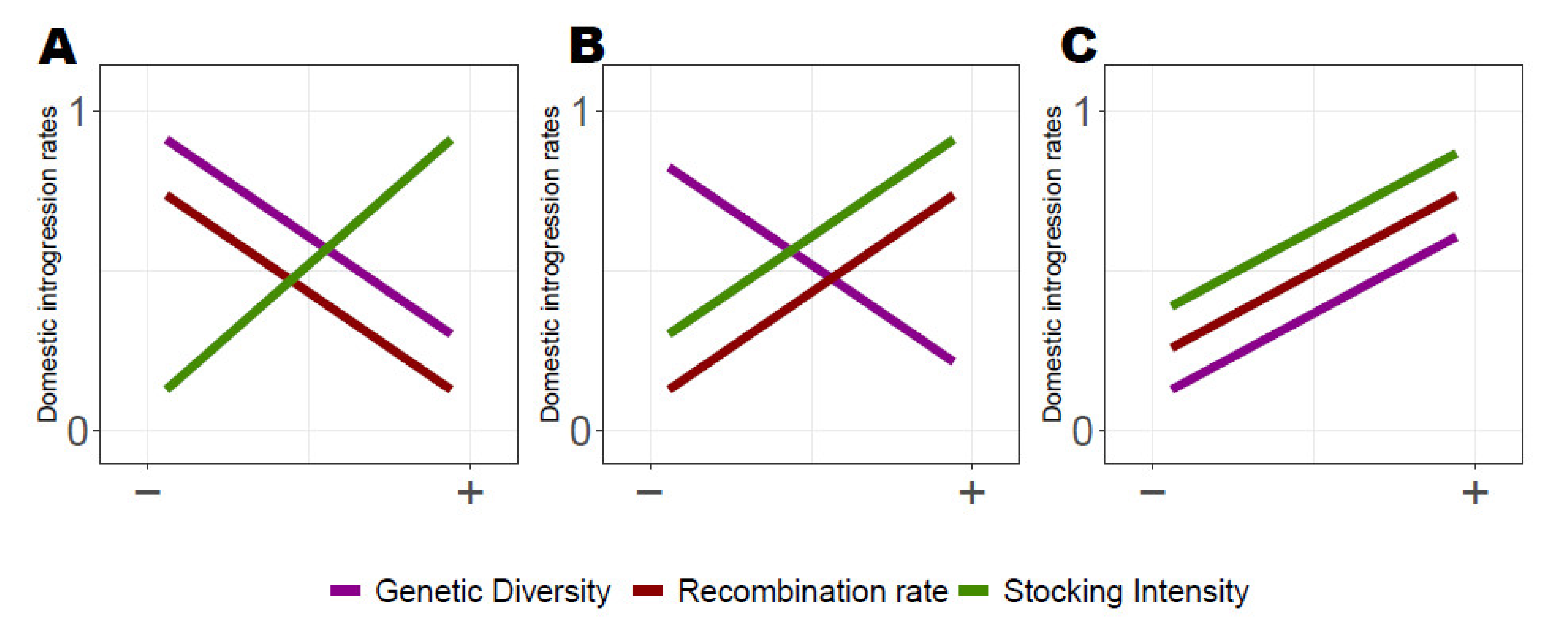
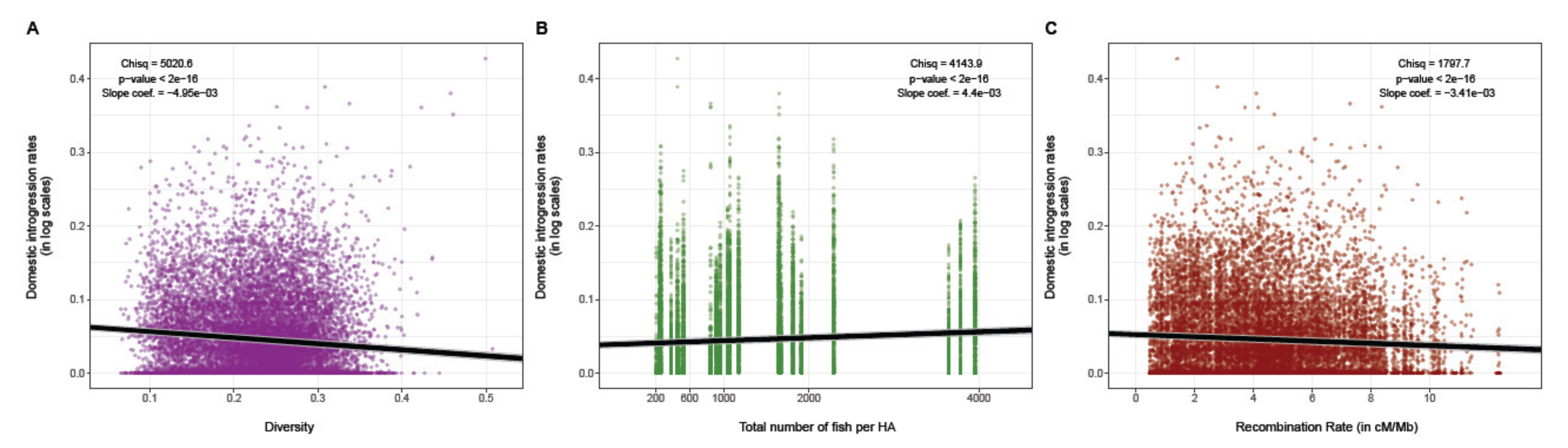
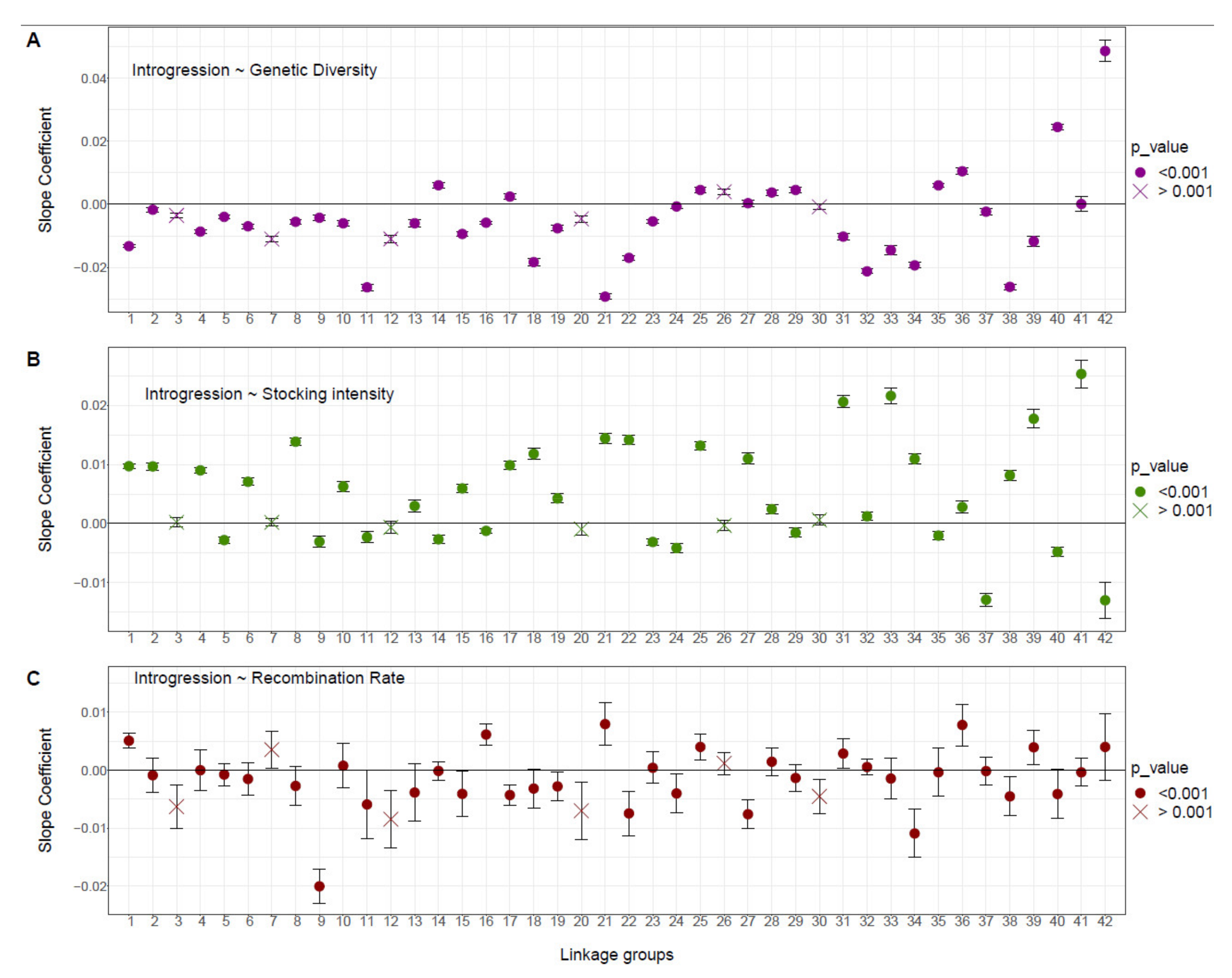
3.2. Influence of Genetic Diversity, Stocking Intensity and Recombination Rate on Domestic Ancestry
4. Discussion
4.1. Associative Overdominance as the Main Mechanism Shaping Domestic Ancestry Landscape
4.2. Negative Epistasis also Contributes to the Dynamics of Domestic Ancestry Landscape
4.3. Perspectives for Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, E. Introgressive Hybridization; John Wiley and Sons, Inc.: New York, NY, USA, 1949. [Google Scholar]
- Dowling, T.E.; Secor, A.C.L. The role of hybridization and introgression in the diversification of animals. Annu. Rev. Ecol. Syst. 1997, 28, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Barton, N.H. The role of hybridization in evolution. Mol. Ecol. 2008, 10, 551–568. [Google Scholar] [CrossRef]
- Harrison, R.G.; Larson, E.L. Hybridization, Introgression, and the Nature of Species Boundaries. J. Hered. 2014, 105, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Racimo, F.; Sankararaman, S.; Nielsen, R.; Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 2015, 16, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindtke, D.; Buerkle, C.A. The genetic architecture of hybrid incompatibilities and their effect on barriers to introgression in secondary contact. Evol. 2015, 69, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Rhymer, J.M.; Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 1996, 27, 83–109. [Google Scholar] [CrossRef]
- Currat, M.; Ruedi, M.; Petit, R.J.; Excoffier, L. The hidden side of invasions: Massive introgression by local genes. Evol. Int. J. Org. Evol. 2008, 62, 1908–1920. [Google Scholar] [CrossRef]
- Knytl, M.; Kalous, L.; Symonova, R.; Rylková, K.; Ráb, P. Chromosome Studies of European Cyprinid Fishes: Cross-Species Painting Reveals Natural Allotetraploid Origin of a Carassius Female with 206 Chromosomes. Cytogenet. Genome Res. 2013, 139, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Knytl, M.; Kalous, L.; Rylková, K.; Choleva, L.; Merilä, J.; Ráb, P. Morphologically indistinguishable hybrid Carassius female with 156 chromosomes: A threat for the threatened crucian carp, C. carassius, L. PLoS ONE 2018, 13, e0190924. [Google Scholar] [CrossRef] [Green Version]
- Laikre, L.; Schwartz, M.K.; Waples, R.S.; Ryman, N.; GeM Working Group. Compromising Genetic Diversity in the Wild: Unmonitored Large-Scale Release of Plants and Animals. Trends Ecol. Evol. 2010, 25, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Hagen, I.J.; Jensen, A.J.; Bolstad, G.H.; Diserud, O.H.; Hindar, K.; Lo, H.; Karlsson, S. Supplementary stocking selects for domesticated genotypes. Nat. Commun. 2019, 10, 199. [Google Scholar] [CrossRef]
- Leitwein, M.; Duranton, M.; Rougemont, Q.; Gagnaire, P.-A.; Bernatchez, L. Using Haplotype Information for Conservation Genomics. Trends Ecol. Evol. 2020, 35, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P.W. Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 2013, 22, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 2015, 24, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.A. The population genetics of speciation: The evolution of hybrid incompatibilities. Genetics 1995, 139, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, S.; Barbash, D.A. The Genetics of Hybrid Incompatibilities. Annu. Rev. Genet. 2011, 45, 331–355. [Google Scholar] [CrossRef] [PubMed]
- Dion-Côté, A.-M.; Renaut, S.; Normandeau, E.; Bernatchez, L. RNA-seq Reveals Transcriptomic Shock Involving Transposable Elements Reactivation in Hybrids of Young Lake Whitefish Species. Mol. Biol. Evol. 2014, 31, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laporte, M.; Le Luyer, J.; Rougeux, C.; Dion-Côté, A.-M.; Krick, M.; Bernatchez, L. DNA methylation reprogramming, TE derepression, and postzygotic isolation of nascent animal species. Sci. Adv. 2019, 5, eaaw1644. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Sankararaman, S.; Mallick, S.; Dannemann, M.; Prüfer, K.; Kelso, J.; Pääbo, S.; Patterson, N.; Reich, D. The genomic landscape of Neanderthal ancestry in present-day humans. Nat. Cell Biol. 2014, 507, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Schumer, M.; Xu, C.; Powell, D.L.; Durvasula, A.; Skov, L.; Holland, C.; Blazier, J.C.; Sankararaman, S.; Andolfatto, P.; Rosenthal, G.G.; et al. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 2018, 360, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.H.; Jiggins, C.D. Interpreting the genomic landscape of introgression. Curr. Opin. Genet. Dev. 2017, 47, 69–74. [Google Scholar] [CrossRef]
- Veller, C.; Edelman, N.B.; Muralidhar, P.; Nowak, M.A. Recombination, Variance in Genetic Relatedness, and Selection against Introgressed DNA. bioRxiv 2019, 846147. [Google Scholar] [CrossRef] [Green Version]
- Lippman, Z.B.; Zamir, D. Heterosis: Revisiting the magic. Trends Genet. 2007, 23, 60–66. [Google Scholar] [CrossRef]
- Chen, Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010, 15, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Medina, P.; Thornlow, B.; Nielsen, R.; Corbett-Detig, R. Estimating the Timing of Multiple Admixture Pulses During Local Ancestry Inference. Genetics 2018, 210, 1089–1107. [Google Scholar] [CrossRef] [Green Version]
- Létourneau, J.; Ferchaud, A.; Le Luyer, J.; Laporte, M.; Garant, D.; Bernatchez, L. Predicting the genetic impact of stocking in Brook Charr (Salvelinus fontinalis) by combining RAD sequencing and modeling of explanatory variables. Evol. Appl. 2017, 11, 577–592. [Google Scholar] [CrossRef]
- White, S.L.; Miller, W.L.; Dowell, S.A.; Bartron, M.L.; Wagner, T. Limited hatchery introgression into wild brook trout (Salvelinus fontinalis ) populations despite reoccurring stocking. Evol. Appl. 2018, 11, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S.J.; Baillie, S.M.; Macmillan, J.; Paterson, I.G.; Buhariwalla, C.F.; Bradbury, I.R.; Bentzen, P. Multiple decades of stocking has resulted in limited hatchery introgression in wild brook trout (Salvelinus fontinalis) populations of Nova Scotia. Evol. Appl. 2020, 13, 1069–1089. [Google Scholar] [CrossRef] [PubMed]
- Leitwein, M.; Cayuela, H.; Ferchaud, A.; Normandeau, É.; Gagnaire, P.; Bernatchez, L. The role of recombination on genome-wide patterns of local ancestry exemplified by supplemented brook charr populations. Mol. Ecol. 2019, 28, 4755–4769. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, F.C.; Sauvage, C.; Marie, A.; Garant, D.; Bernatchez, L. Dynamics of introgressive hybridization assessed by SNP population genomics of coding genes in stocked brook charr (Salvelinus fontinalis). Mol. Ecol. 2012, 21, 2877–2895. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.Y.; Huber, C.D.; Lohmueller, K.E. Deleterious variation shapes the genomic landscape of introgression. PLoS Genet. 2018, 14, e1007741. [Google Scholar] [CrossRef] [Green Version]
- Schumer, M.; Rosenthal, G.G.; Andolfatto, P. How common is homoploid hybrid speciation? Evolution 2014, 68, 1553–1560. [Google Scholar] [CrossRef]
- Duranton, M.; Allal, F.; Fraïsse, C.; Bierne, N.; Bonhomme, F.; Gagnaire, P.-A. The origin and remolding of genomic islands of differentiation in the European sea bass. Nat. Commun. 2018, 9, 2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.H.; Davey, J.W.; Salazar, C.; Jiggins, C.D. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLoS Biol. 2019, 17, e2006288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ministère du Développement Durable, de l’Environnement, de la Faune et des Parcs. Outil D’aide À L’ensemencement Des Plans D’eau—Omble de Fontaine (Salvelinus Fontinalis). Direction de La Faune Aquatique: Québec, Canada; Direction de La Faune Aquatique: Québec, Canada; Direction Générale de l’expertise Sur La Faune et Ses Habitats: Québec, Canada, 2013; pp. 1–12.
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.A.; Rondeau, E.B.; Minkley, D.R.; Leong, J.S.; Nugent, C.M.; Danzmann, R.G.; Ferguson, M.M.; Stadnik, A.; Devlin, R.H.; Muzzerall, R.; et al. The Arctic charr (Salvelinus alpinus) genome and transcriptome assembly. PLoS ONE 2018, 13, e0204076. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinform. 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, T.; Bernatchez, L. Stackr: GBS/RAD Data Exploration, Manipulation and Visualization Using R. 2016. Available online: Https://Github.Com/Thierrygosselin/Stackr (accessed on 22 March 2021).
- Sutherland, B.J.G.; Gosselin, T.; Normandeau, E.; Lamothe, M.; Isabel, N.; Audet, C.; Bernatchez, L. Salmonid chromosome evolution as revealed by a novel method for comparing RADseq linkage maps. Genome Biol. Evol. 2016, 8, 3600–3617. [Google Scholar] [CrossRef]
- Guan, Y. Detecting Structure of Haplotypes and Local Ancestry. Genetics 2014, 196, 625–642. [Google Scholar] [CrossRef] [Green Version]
- Leitwein, M.; Gagnaire, P.-A.; Desmarais, E.; Berrebi, P.; Guinand, B. Genomic consequences of a recent three-way admixture in supplemented wild brown trout populations revealed by local ancestry tracts. Mol. Ecol. 2018, 27, 3466–3483. [Google Scholar] [CrossRef] [PubMed]
- Brasil República. Decreto—Lei nº 227, de 28 de fevereiro de 1967. Dá nova redação ao Decreto-lei nº 1.985, de 29 de janeiro de 1940 (Código de Minas). 1967. Available online: http://www.planalto.gov.br/ccivil_03/Decreto-Lei/Del0227.htm (accessed on 19 October 2020).
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvoy, C.; Charif, D.; Guéguen, L.; Marais, G.A. MareyMap: An R-based tool with graphical interface for estimating recombination rates. Bioinformatics 2007, 23, 2188–2189. [Google Scholar] [CrossRef] [PubMed]
- Etymologia: Bonferroni Correction. Emerg. Infect. Dis. 2015, 21, 289. [CrossRef]
- Todesco, M.; Pascual, M.A.; Owens, G.L.; Ostevik, K.L.; Moyers, B.T.; Hübner, S.; Heredia, S.M.; Hahn, M.A.; Caseys, C.; Bock, D.G.; et al. Hybridization and extinction. Evol. Appl. 2016, 9, 892–908. [Google Scholar] [CrossRef]
- Marie, A.D.; Bernatchez, L.; Garant, D. Loss of genetic integrity correlates with stocking intensity in brook charr (Salvelinus fontinalis). Mol. Ecol. 2010, 19, 2025–2037. [Google Scholar] [CrossRef]
- Luikart, G.; Ryman, N.; Tallmon, D.A.; Schwartz, M.K.; Allendorf, F.W. Estimation of census and effective population sizes: The increasing usefulness of DNA-based approaches. Conserv. Genet. 2010, 11, 355–373. [Google Scholar] [CrossRef]
- Gossieaux, P.; Bernatchez, L.; Sirois, P.; Garant, D. Impacts of stocking and its intensity on effective population size in Brook Charr (Salvelinus fontinalis) populations. Conserv. Genet. 2019, 20, 729–742. [Google Scholar] [CrossRef]
- Ferchaud, A.; Leitwein, M.; Laporte, M.; Boivin-Delisle, D.; Bougas, B.; Hernandez, C.; Normandeau, É.; Thibault, I.; Bernatchez, L. Adaptive and maladaptive genetic diversity in small populations: Insights from the Brook Charr (Salvelinus fontinalis) case study. Mol. Ecol. 2020, 29, 3429–3445. [Google Scholar] [CrossRef]
- Richards, C.M. Inbreeding Depression and Genetic Rescue in a Plant Metapopulation. Am. Nat. 2000, 155, 383–394. [Google Scholar] [CrossRef]
- Uller, T.; Leimu, R. Founder events predict changes in genetic diversity during human-mediated range expansions. Glob. Chang. Biol. 2011, 17, 3478–3485. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Garcia-Dorado, A. Understanding Inbreeding Depression, Purging, and Genetic Rescue. Trends Ecol. Evol. 2016, 31, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. What is the evidence for heterozygote advantage selection? Trends Ecol. Evol. 2012, 27, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Zhang, Y.; Nielsen, R. Genetic rescue and the maintenance of native ancestry. Conserv. Genet. 2019, 20, 59–64. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef]
- Schou, M.F.; Loeschcke, V.; Bechsgaard, J.; Schlötterer, C.; Kristensen, T.N. Unexpected high genetic diversity in small populations suggests maintenance by associative overdominance. Mol. Ecol. 2017, 26, 6510–6523. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.J.; Pouyet, F.; Excoffier, L.; Peischl, S. Transition from Background Selection to Associative Overdominance Promotes Diversity in Regions of Low Recombination. Curr. Biol. 2020, 30, 101–107. [Google Scholar] [CrossRef]
- Turelli, M.; Orr, H.A. Dominance, epistasis and the genetics of postzygotic isolation. Genetics 2000, 154, 1663–1679. [Google Scholar]
- Orr, H.A.; Turelli, M. The evolution of postzygotic isolation: Accumulating Dobzhansky-Muller incompatibilities. Evol. Int. J. Org. Evol. 2001, 55, 1085–1094. [Google Scholar] [CrossRef]
- Renaut, S.; Bernatchez, L. Transcriptome-wide signature of hybrid breakdown associated with intrinsic reproductive isolation in lake whitefish species pairs (Coregonus spp. Salmonidae). Heredity 2010, 106, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R. Genetic adaptation to captivity in species conservation programs. Mol. Ecol. 2008, 17, 325–333. [Google Scholar] [CrossRef]
- Bosse, M.; Megens, H.; Derks, M.F.L.; De Cara, M.; Ángeles, R.; Groenen, M.A.M. Deleterious alleles in the context of domestication, inbreeding, and selection. Evol. Appl. 2018, 12, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.B.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the Probability of Outbreeding Depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef]
- Marshall, T.C.; Spalton, J.A. Simultaneous inbreeding and outbreeding depression in reintroduced Arabian oryx. Anim. Conserv. 2000, 3, 241–248. [Google Scholar] [CrossRef]
- Frankham, R. Where are we in conservation genetics and where do we need to go? Conserv. Genet. 2009, 11, 661–663. [Google Scholar] [CrossRef]
- Huff, D.D.; Miller, L.M.; Chizinski, C.J.; Vondracek, B. Mixed-source reintroductions lead to outbreeding depression in second-generation descendents of a native North American fish. Mol. Ecol. 2011, 20, 4246–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaulfuß, F.; Reisch, C. Reintroduction of the endangered and endemic plant species Cochlearia bavarica—Implications from conservation genetics. Ecol. Evol. 2017, 7, 11100–11112. [Google Scholar] [CrossRef] [PubMed]
- Mable, B.K. Conservation of adaptive potential and functional diversity: Integrating old and new approaches. Conserv. Genet. 2019, 20, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.C.A.; Deloukas, P.; Hunt, S.; Mullikin, J.; Myers, S.; Silverman, B.; Donnelly, P.; Bentley, D.; McVean, G. The Influence of Recombination on Human Genetic Diversity. PLoS Genet. 2006, 2, e148. [Google Scholar] [CrossRef]
- Burke, M.K. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc. R. Soc. B Biol. Sci. 2012, 279, 5029–5038. [Google Scholar]
- Bougas, B.; Granier, S.; Audet, C.; Bernatchez, L. The Transcriptional Landscape of Cross-Specific Hybrids and Its Possible Link with Growth in Brook Charr (Salvelinus fontinalis Mitchill). Genetics 2010, 186, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Cook, C.N.; Sgrò, C.M. Poor understanding of evolutionary theory is a barrier to effective conservation management. Conserv. Lett. 2019, 12, 12619. [Google Scholar] [CrossRef]
- Harris, K.; Nielsen, R. The Genetic Cost of Neanderthal Introgression. Genetics 2016, 203, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reserve | Lake | Label | N_samples_filters | N_late_hybrids | N_SNPs_mapped | lakes_size | total_ha | Mean Ancestry Rate | Mean Genetic Diversity |
|---|---|---|---|---|---|---|---|---|---|
| Mastigouche | Abénakis | ABE | 21 | 16 | 31529 | 5 | 1915 | 0.0253 | 0.259 |
| Arbout | ARB | 28 | 24 | 31713 | 5 | 380 | 0.0261 | 0.259 | |
| Chamberlain | CHA | 20 | 19 | 33290 | 18 | 958 | 0.0324 | 0.261 | |
| Cougouar | COU | 29 | 28 | 33455 | 8 | 1669 | 0.0426 | 0.252 | |
| Deux-Etapes | DET | 28 | 22 | 33580 | 12.3 | 3647 | 0.0282 | 0.246 | |
| Gélinotte | GEL | 25 | 18 | 31377 | 5 | 1652 | 0.0209 | 0.271 | |
| Grignon | GRI | 23 | 17 | 33298 | 29.6 | 846 | 0.0186 | 0.26 | |
| Jones | JON | 26 | 17 | 30553 | 28 | 468 | 0.0062 | 0.25 | |
| Ledoux | LED | 26 | 12 | 30941 | 13.7 | 3956 | 0.0663 | 0.236 | |
| Lemay | LEM | 28 | 19 | 30253 | 19.1 | 914 | 0.0538 | 0.23 | |
| Saint-Maurice | Brulȏt | BRU | 25 | 17 | 35463 | 8.1 | 247 | 0.0557 | 0.166 |
| Corbeil | COR | 23 | 16 | 28071 | 9.5 | 526 | 0.0491 | 0.261 | |
| Gaspard | GAS | 28 | 14 | 34844 | 11.6 | 259 | 0.0766 | 0.168 | |
| Maringouins | MAR | 26 | 17 | 31102 | 6.2 | 1073 | 0.0734 | 0.23 | |
| Melchior | MEL | 26 | 20 | 30720 | 4.4 | 455 | 0.038 | 0.212 | |
| Milord | MIL | 25 | 16 | 41035 | 46.7 | 1175 | 0.0843 | 0.158 | |
| Perdu | PER | 24 | 17 | 32484 | 22.1 | 2293 | 0.075 | 0.247 | |
| Porc-Epic | POE | 26 | 17 | 32100 | 2.7 | 1648 | 0.0829 | 0.247 | |
| Portage | POR | 27 | 12 | 34415 | 46.9 | 1044 | 0.0615 | 0.157 | |
| Tempȇte | TEM | 17 | 11 | 32852 | 12.5 | 3784 | 0.046 | 0.286 | |
| À la truite | TRU | 26 | 21 | 31741 | 6.5 | 1814 | 0.0597 | 0.26 | |
| Vierge | VIE | 26 | 12 | 33747 | 5.7 | 208 | 0.0052 | 0.176 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitwein, M.; Cayuela, H.; Bernatchez, L. Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations. Genes 2021, 12, 524. https://doi.org/10.3390/genes12040524
Leitwein M, Cayuela H, Bernatchez L. Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations. Genes. 2021; 12(4):524. https://doi.org/10.3390/genes12040524
Chicago/Turabian StyleLeitwein, Maeva, Hugo Cayuela, and Louis Bernatchez. 2021. "Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations" Genes 12, no. 4: 524. https://doi.org/10.3390/genes12040524
APA StyleLeitwein, M., Cayuela, H., & Bernatchez, L. (2021). Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations. Genes, 12(4), 524. https://doi.org/10.3390/genes12040524