
A Link between Replicative Stress, Lamin Proteins, and Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
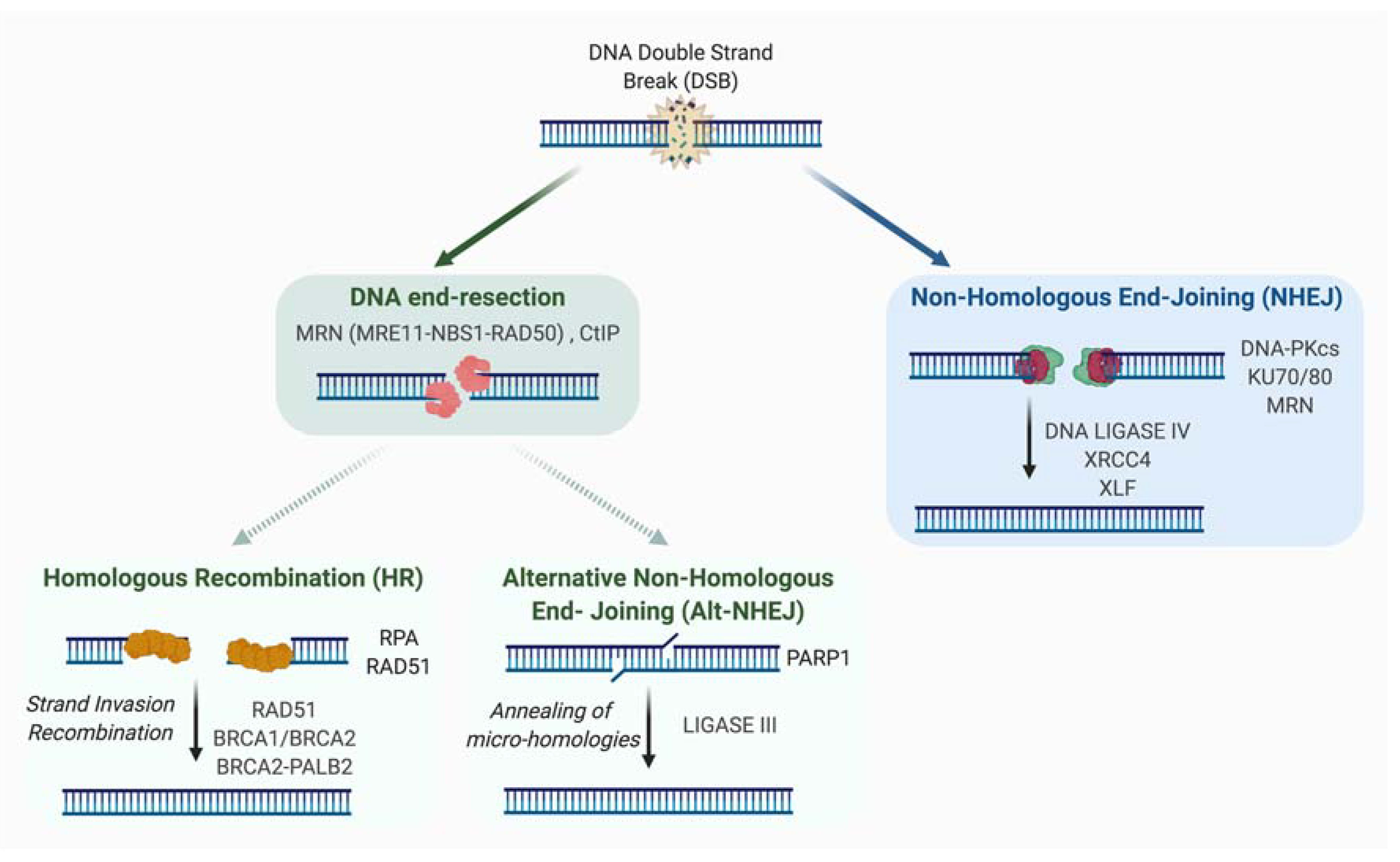
2. Mechanisms of Double-Strand Break Repair
2.1. Non-Homologous End-Joining (NHEJ)
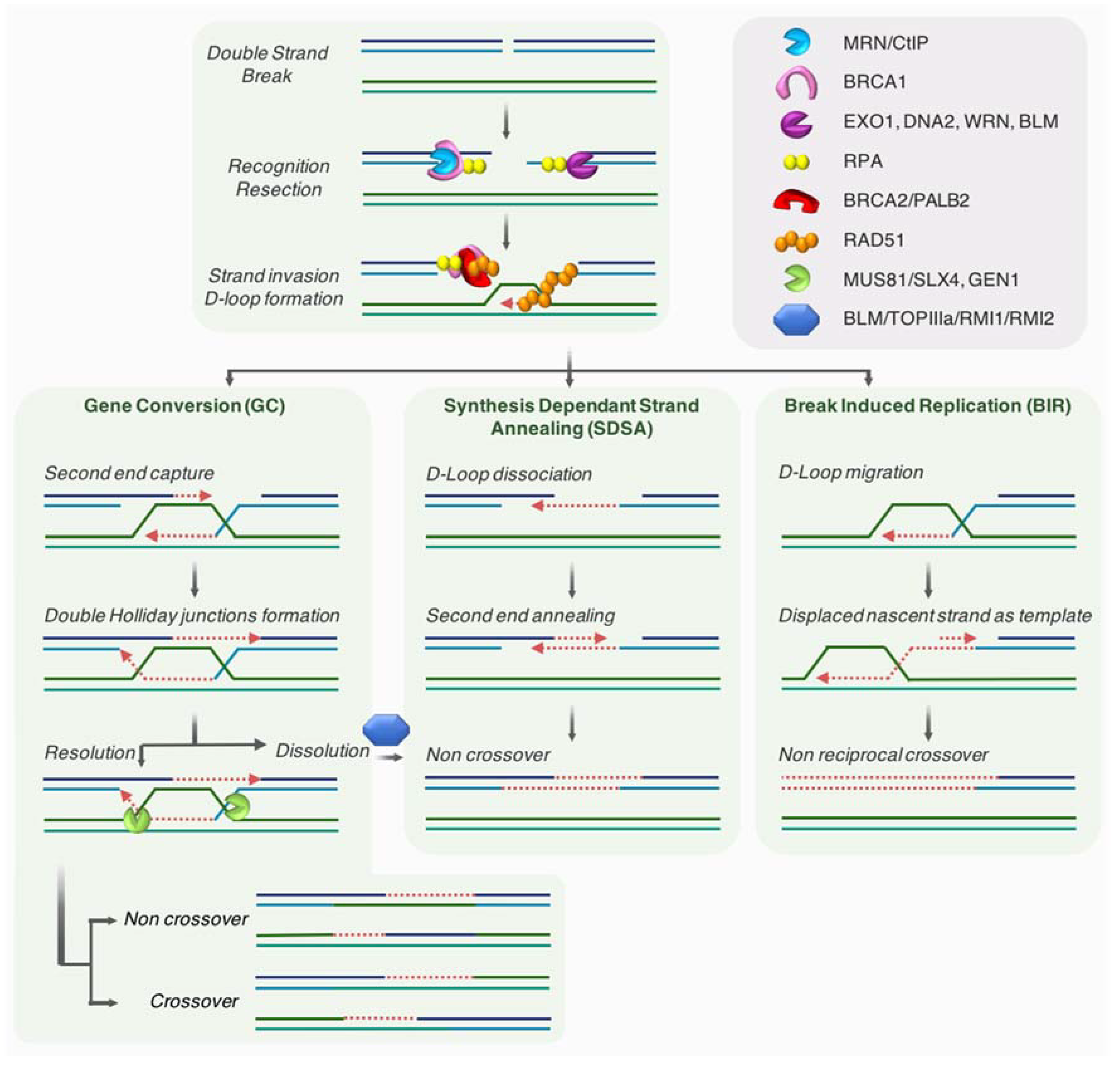
2.2. Homologous Recombination
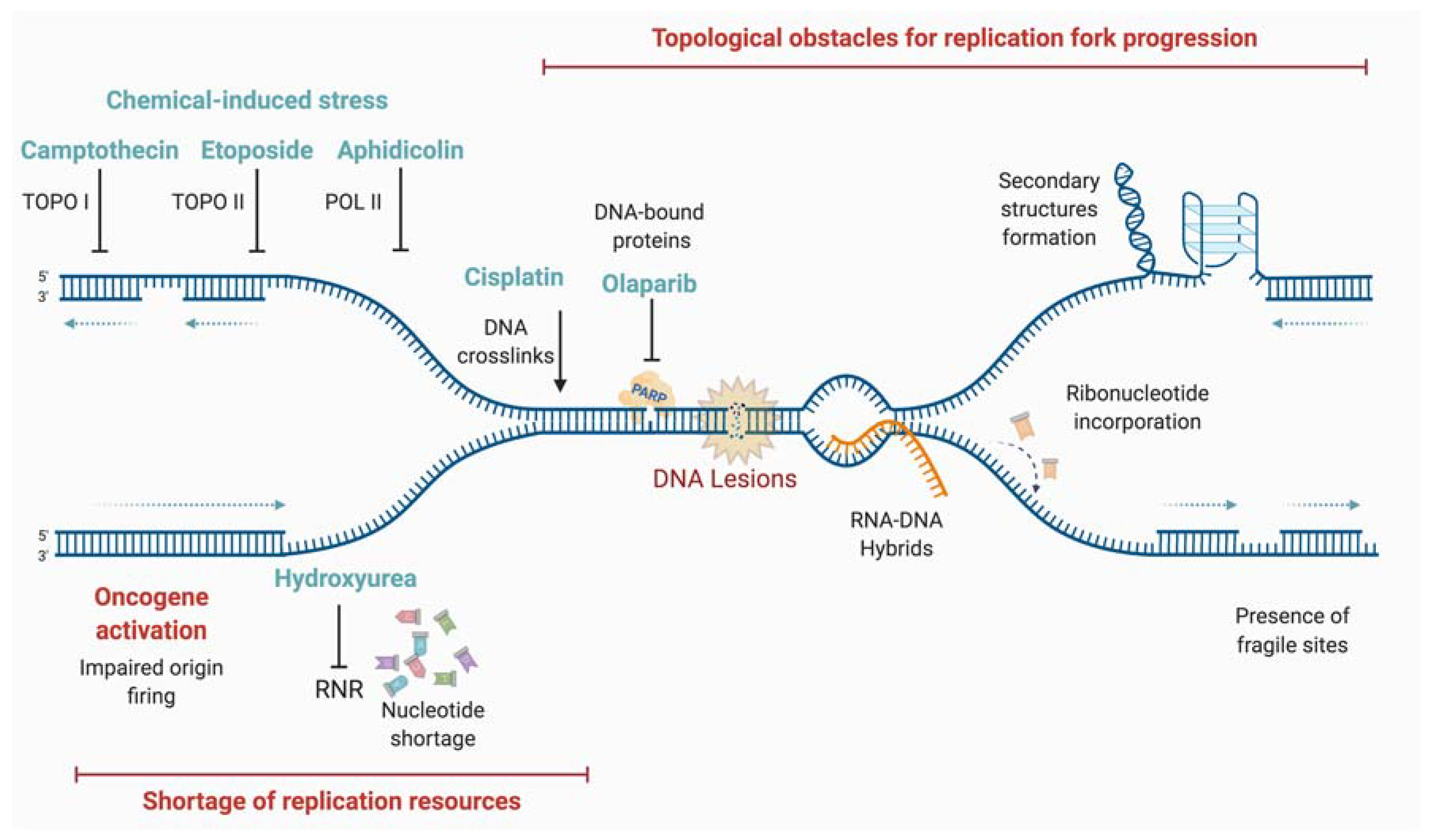
3. Replicative Stress
3.1. Different Causes of Replicative Stress
3.1.1. Oxidative Lesions: “Natural” Causes of Replicative Stress
3.1.2. Repetitive Sequences and Particular DNA Structures
3.1.3. RNA-DNA Hybrids
3.1.4. Oncogenes
3.1.5. Different Lesions
3.1.6. Defective Replication Process
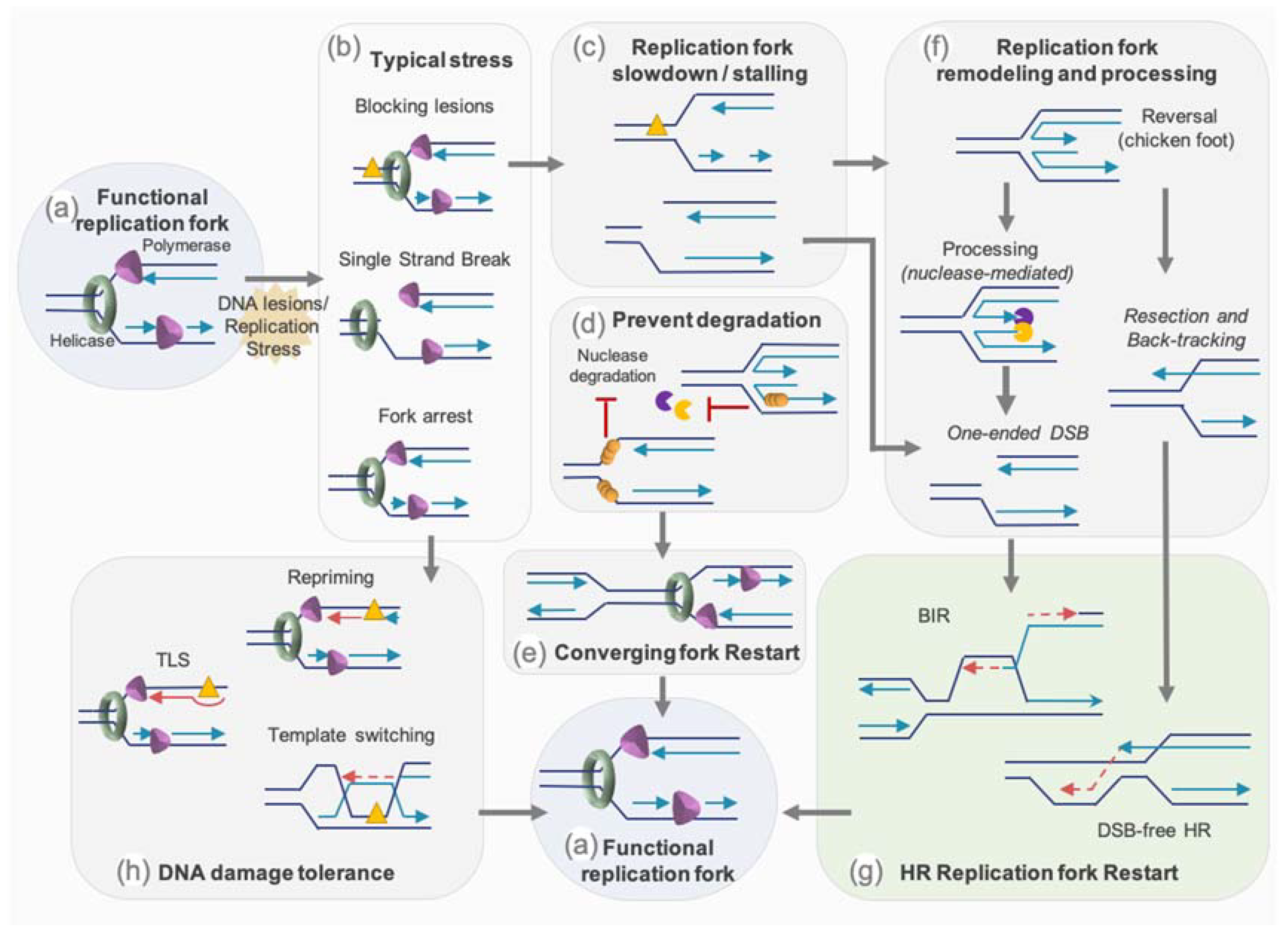
3.2. How Is Replicative Stress Managed?
3.2.1. Avoiding New Origin Firing—Role of ATR
3.2.2. Restart by Homology-Mediated Recombination
3.2.3. Protection of Stalled or Reversed Forks
3.2.4. Nucleases and Helicases Mediating HR Restart of the Fork
3.2.5. Translesion Synthesis and Repriming
3.3. Responses to Avoid Genome Instability upon Replicative Stress
3.3.1. Nature of DSB during Replicative Stress and the Role of Sister Chromatid Cohesion
3.3.2. Mechanisms to Avoid Replication Stress-Induced Chromosome Aberrations by Precluding NHEJ Factor or by Positively Regulating HR
3.3.3. Consequences during Mitosis
3.4. Consequence of Replicative Stress on Inflammation
4. Presentation of Lamins
4.1. Two Types of Mammalian Lamins
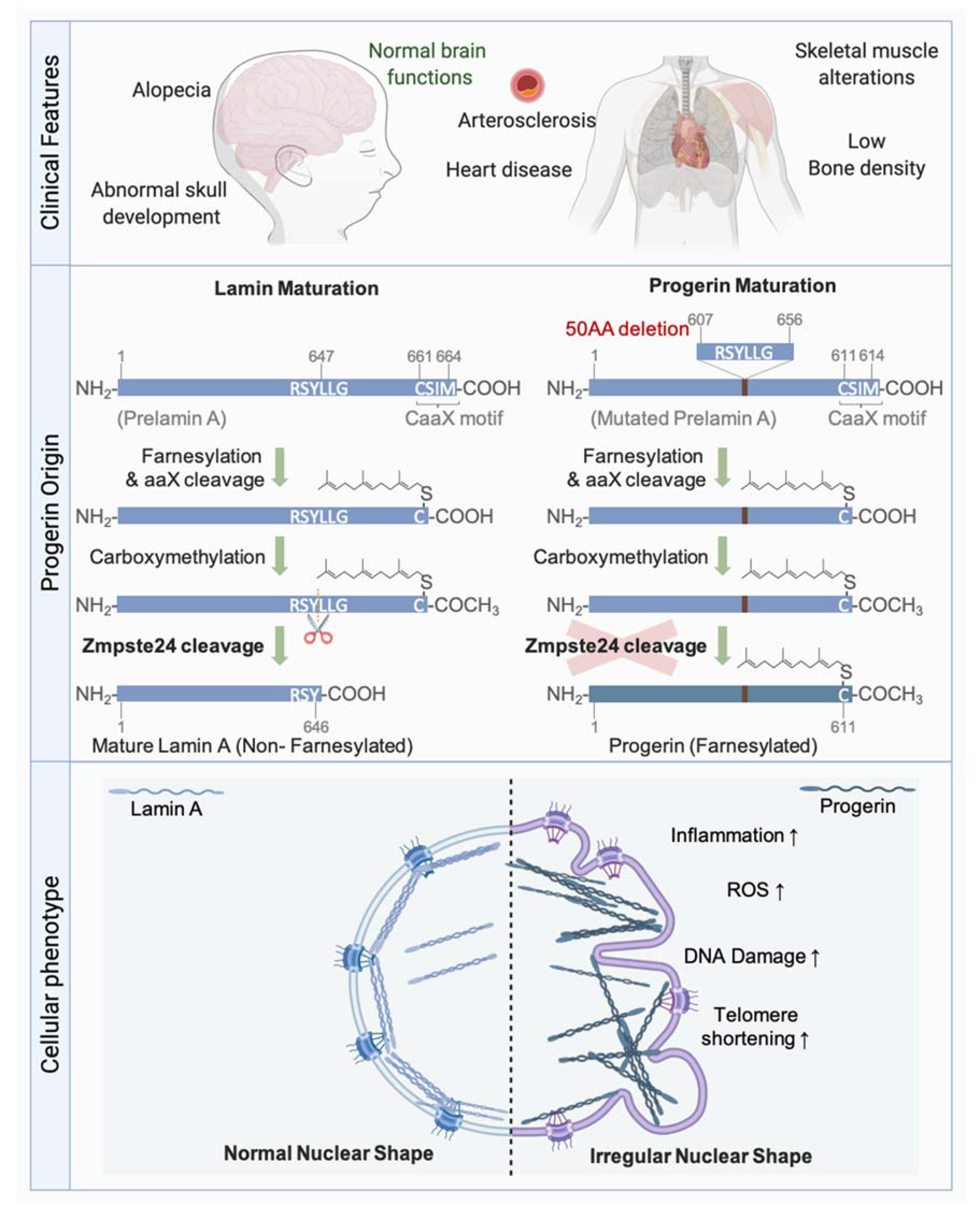
4.2. Maturation of Lamins
4.3. Associated Pathologies
4.4. Lamins and Senescence
4.4.1. A-Type Lamins and Senescence
4.4.2. B-Type Lamins and Senescence
4.5. Lamins and Cancer
5. Lamins and Genome Stability: Their Roles during DNA Repair, Replication, or Replicative Stress
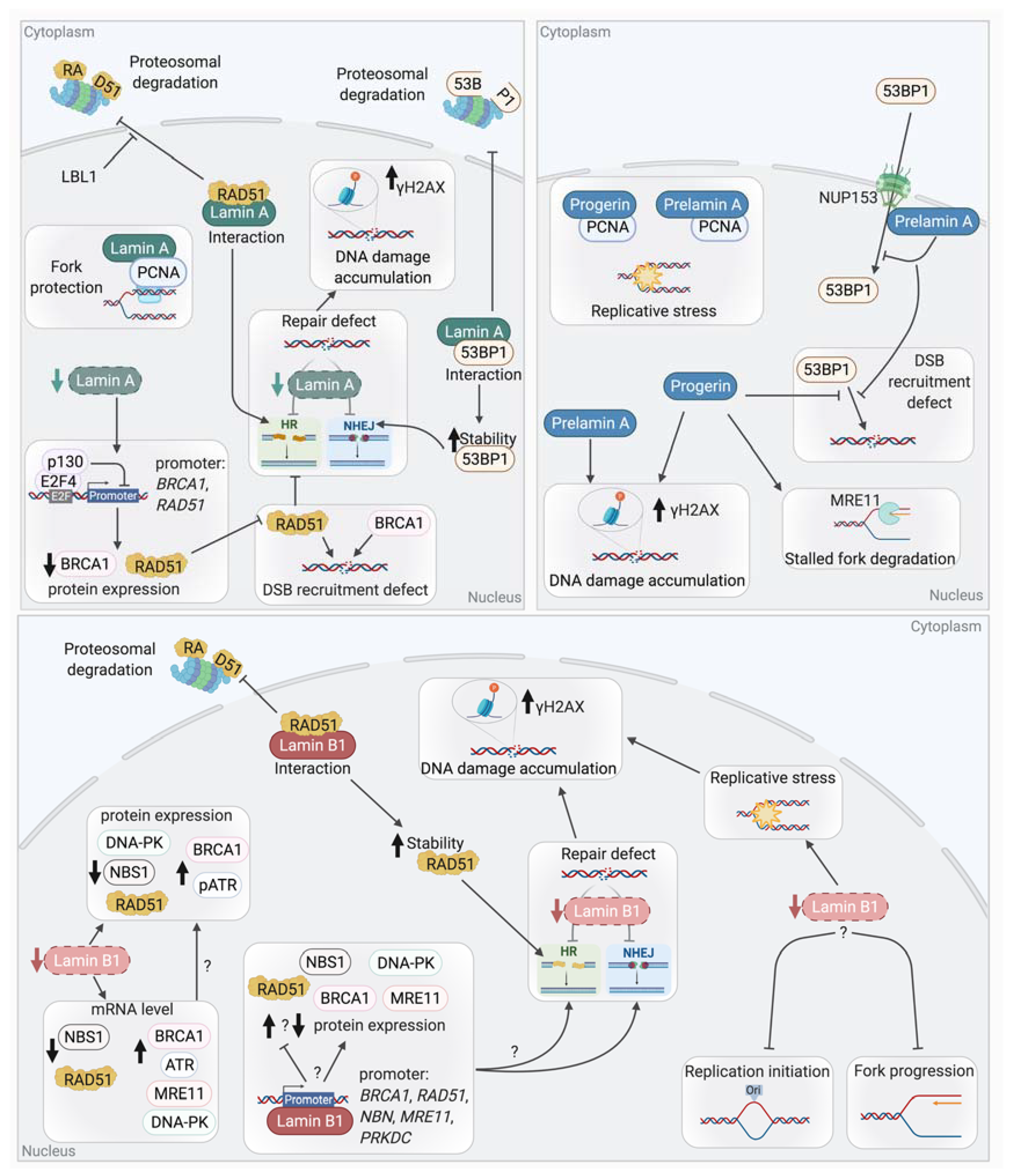
5.1. Lamins and DNA Repair
5.1.1. Lamin A, Chromatin Modifications, and DNA Damage
5.1.2. Lamin A and NHEJ
5.1.3. Lamin A and Homologous Recombination
5.1.4. Lamin B1 and DSB Repair
5.1.5. Lamins and Other DNA Repair Mechanisms
5.2. Lamins and Telomere Maintenance
5.3. Lamins, Replication, and Replicative Stress
5.3.1. On Replication Progression
5.3.2. Replication Timing
5.3.3. During Replicative Stress
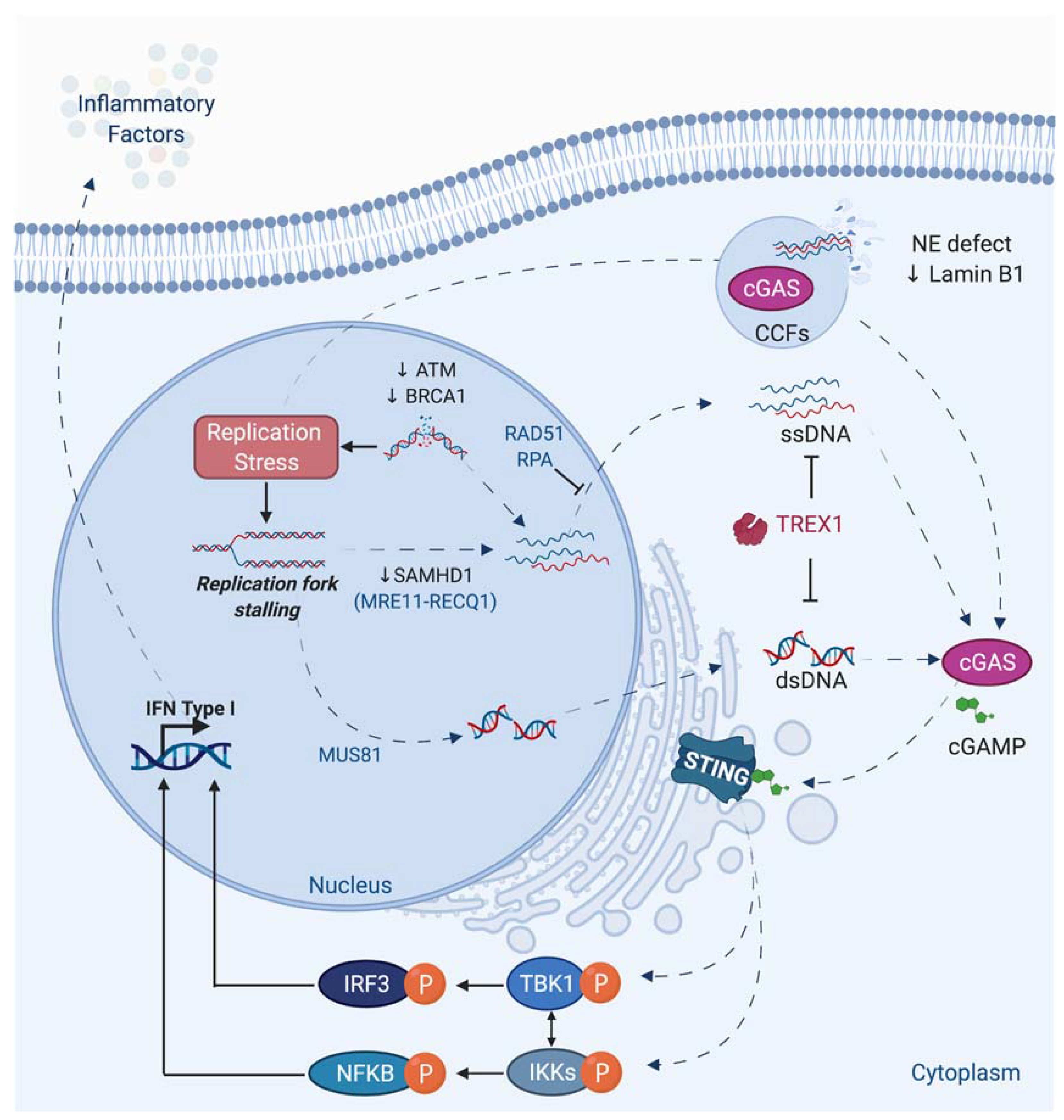
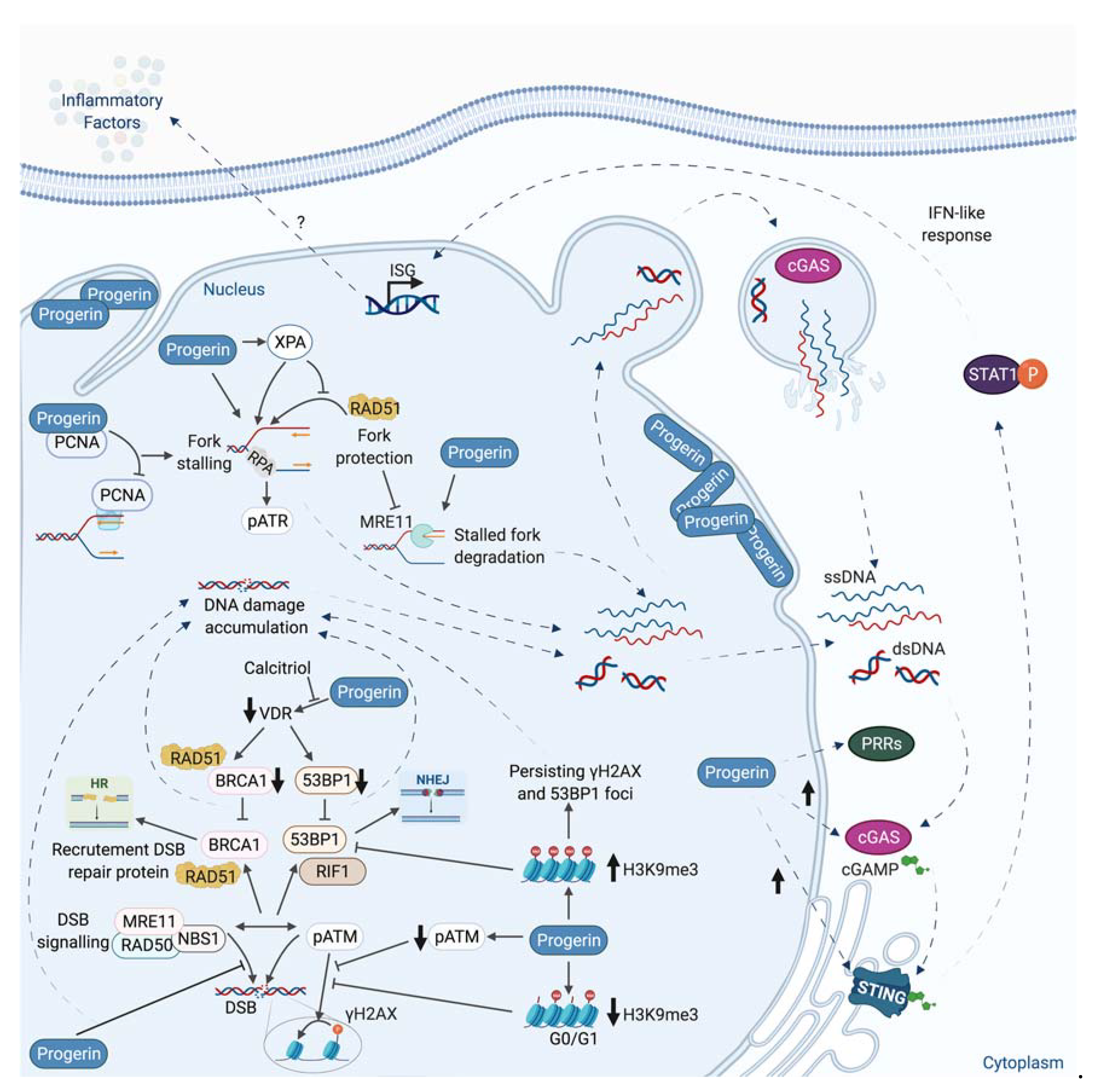
6. A Link between Nuclear Envelope Integrity, DNA Damage, Inflammation, and Aging: The Case of the HGP Syndrome
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berti, M.; Vindigni, M.B.A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The Plasticity of DNA Replication Forks in Response to Clinically Relevant Genotoxic Stress. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Pasero, P.; Vindigni, A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [Green Version]
- Coquel, F.; Neumayer, C.; Lin, Y.-L.; Pasero, P. SAMHD1 and the Innate Immune Response to Cytosolic DNA during DNA Replication. Curr. Opin. Immunol. 2019, 56, 24–30. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through cGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M. Nuclear Envelope Rupture: Little Holes, Big Openings. Curr. Opin. Cell Biol. 2018, 52, 66–72. [Google Scholar] [CrossRef]
- Gonzalo, S.; Coll-Bonfill, N. Genomic Instability and Innate Immune Responses to Self-DNA in Progeria. GeroScience 2019, 41, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.-C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Duboc, C.; Wu, Q.; Ochi, T.; Liang, S.; Tsutakawa, S.E.; Lees-Miller, S.P.; Nadal, M.; Tainer, J.A.; Blundell, T.L.; et al. Dissection of DNA Double-Strand-Break Repair Using Novel Single-Molecule Forceps. Nat. Struct. Mol. Biol. 2018, 25, 482–487. [Google Scholar] [CrossRef]
- Graham, T.G.W.; Walter, J.C.; Loparo, J.J. Two-Stage Synapsis of DNA Ends during Non-Homologous End Joining. Mol. Cell 2016, 61, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.A.; Keegan, S.; Leo-Macias, A.; Watanabe, G.; Strande, N.T.; Chang, H.H.; Oksuz, B.A.; Fenyo, D.; Lieber, M.R.; Ramsden, D.A.; et al. Organization and Dynamics of the Nonhomologous End-Joining Machinery during DNA Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2015, 112, E2575–E2584. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Watanabe, G.; Morten, M.J.; Reid, D.A.; Rothenberg, E.; Lieber, M.R. The Essential Elements for the Noncovalent Association of Two DNA Ends during NHEJ Synapsis. Nat. Commun. 2019, 10, 3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondaneche, M.; Sanal, O.; Plebani, A.; Stephan, J.; Hufnagel, M.; le Deist, F.; et al. Cernunnos, a Novel Nonhomologous End-Joining Factor, Is Mutated in Human Immunodeficiency with Microcephaly. Cell 2006, 124, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Riballo, E.; Woodbine, L.; Stiff, T.; Walker, S.; Goodarzi, A.; Jeggo, P. XLF-Cernunnos Promotes DNA Ligase IV-XRCC4 Re-Adenylation Following Ligation. Nucleic Acids Res. 2009, 37, 482–492. [Google Scholar] [CrossRef]
- Tadi, S.K.; Tellier-Lebègue, C.; Nemoz, C.; Drevet, P.; Audebert, S.; Roy, S.; Meek, K.; Charbonnier, J.-B.; Modesti, M. PAXX Is an Accessory c-NHEJ Factor That Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep. 2016, 17, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. DNA Repair. PAXX, a Paralog of XRCC4 and XLF, Interacts with Ku to Promote DNA Double-Strand Break Repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, I.; Sitters, G.; Candelli, A.; Heerema, S.J.; Heller, I.; de Melo, A.J.; Zhang, H.; Normanno, D.; Modesti, M.; Peterman, E.J.G.; et al. Sliding Sleeves of XRCC4-XLF Bridge DNA and Connect Fragments of Broken DNA. Nature 2016, 535, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Ropars, V.; Drevet, P.; Legrand, P.; Baconnais, S.; Amram, J.; Faure, G.; Márquez, J.A.; Piétrement, O.; Guerois, R.; Callebaut, I.; et al. Structural Characterization of Filaments Formed by Human Xrcc4-Cernunnos/XLF Complex Involved in Nonhomologous DNA End-Joining. Proc. Natl. Acad. Sci. USA 2011, 108, 12663–12668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, S.N.; Vergnes, A.; Ristic, D.; Wyman, C.; Modesti, M.; Junop, M. A Human XRCC4-XLF Complex Bridges DNA. Nucleic Acids Res. 2012, 40, 1868–1878. [Google Scholar] [CrossRef] [Green Version]
- Hammel, M.; Rey, M.; Yu, Y.; Mani, R.S.; Classen, S.; Liu, M.; Pique, M.E.; Fang, S.; Mahaney, B.L.; Weinfeld, M.; et al. XRCC4 Protein Interactions with XRCC4-like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair. J. Biol. Chem. 2011, 286, 32638–32650. [Google Scholar] [CrossRef] [Green Version]
- Nemoz, C.; Ropars, V.; Frit, P.; Gontier, A.; Drevet, P.; Yu, J.; Guerois, R.; Pitois, A.; Comte, A.; Delteil, C.; et al. XLF and APLF Bind Ku80 at Two Remote Sites to Ensure DNA Repair by Non-Homologous End Joining. Nat. Struct. Mol. Biol. 2018, 25, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The Molecular Basis and Disease Relevance of Non-Homologous DNA End Joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 Pathway on NHEJ-Induced Genome Rearrangements in Mammalian Cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.; Rass, E.; Plo, I.; Bertrand, P.; Lopez, B. Defects in XRCC4 and KU80 Differentially Affect the Joining of Distal Nonhomologous Ends. Proc. Natl. Acad. Sci. USA 2007, 104, 20902–20907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B. Role of Mre11 in Chromosomal Nonhomologous End Joining in Mammalian Cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Guirouilh-Barbat, J.; Barascu, A.; Pennarun, G.; Genet, D.; Rass, E.; Germann, S.; Bertrand, P.; Hickson, I.; Lopez, B. A Role for BLM in Double-Strand Break Repair Pathway Choice: Prevention of CtIP/Mre11-Mediated Alternative Nonhomologous End-Joining. Cell Rep. 2013, 5, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betermier, M.; Bertrand, P.; Lopez, B. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Perrault, A.R.; Takeda, Y.; Qin, W.; Iliakis, G. Biochemical Evidence for Ku-Independent Backup Pathways of NHEJ. Nucleic Acids Res. 2003, 31, 5377–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA Ligase III as a Candidate Component of Backup Pathways of Nonhomologous End Joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku Compete for Repair of DNA Double Strand Breaks by Distinct NHEJ Pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-Ribose) Polymerase-1 and XRCC1/DNA Ligase III in an Alternative Route for DNA Double-Strand Breaks Rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [Green Version]
- Xie, A.; Kwok, A.; Scully, R. Role of Mammalian Mre11 in Classical and Alternative Nonhomologous End Joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Grabarz, A.; Barascu, A.; Guirouilh-Barbat, J.; Lopez, B. Initiation of DNA Double Strand Break Repair: Signaling and Single-Stranded Resection Dictate the Choice between Homologous Recombination, Non-Homologous End-Joining and Alternative End-Joining. Am. J. Cancer Res. 2012, 2, 249–268. [Google Scholar]
- Beck, C.; Boehler, C.; Guirouilh Barbat, J.; Bonnet, M.; Illuzzi, G.; Ronde, P.; Gauthier, L.; Magroun, N.; Rajendran, A.; Lopez, B.; et al. PARP3 Affects the Relative Contribution of Homologous Recombination and Nonhomologous End-Joining Pathways. Nucleic Acids Res. 2014, 42, 5616–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Cejka, P. DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J. Biol. Chem. 2015, 290, 22931–22938. [Google Scholar] [CrossRef] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sun, H.; Huang, Y.; Wang, Y.; Liu, Y.; Chen, X. Pathways and assays for DNA double-strand break repair by homologous recombination. Acta Biochim. Biophys. Sin. 2019, 51, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main Steps in DNA Double-Strand Break Repair: An Introduction to Homologous Recombination and Related Processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B. Is Homologous Recombination Really an Error-Free Process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakofsky, C.J.; Malkova, A. Break Induced Replication in Eukaryotes: Mechanisms, Functions, and Consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-Induced Replication Repair of Damaged Forks Induces Genomic Duplications in Human Cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Roumelioti, F.-M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative Lengthening of Human Telomeres Is a Conservative DNA Replication Process with Features of Break-Induced Replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-Induced Telomere Synthesis Underlies Alternative Telomere Maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Takai, K.K.; Lovejoy, C.A.; de Lange, T. Break-Induced Replication Promotes Fragile Telomere Formation. Genes Dev. 2020, 34, 1392–1405. [Google Scholar] [CrossRef]
- Carr, A.M.; Lambert, S. Replication Stress-Induced Genome Instability: The Dark Side of Replication Maintenance by Homologous Recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving Replication Fork Integrity and Competence via the Homologous Recombination Pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, K.; Smogorzewska, A. Advances in Understanding DNA Processing and Protection at Stalled Replication Forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic Signaling Mediated by Oxidants in Ras-Transformed Fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, S.S. Biological Consequences of Free Radical-Damaged DNA Bases. Free Radic. Biol. Med. Vol. 2002, 33, 1–14. [Google Scholar] [CrossRef]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [Green Version]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-Mediated Replication Fork Reversal Is a Global Response to Genotoxic Treatments in Human Cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Somyajit, K.; Gupta, R.; Sedlackova, H.; Neelsen, K.J.; Ochs, F.; Rask, M.-B.; Choudhary, C.; Lukas, J. Redox-Sensitive Alteration of Replisome Architecture Safeguards Genome Integrity. Science 2017, 358, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Aller, P.; Rould, M.A.; Hogg, M.; Wallace, S.S.; Doublié, S. A Structural Rationale for Stalling of a Replicative DNA Polymerase at the Most Common Oxidative Thymine Lesion, Thymine Glycol. Proc. Natl. Acad. Sci. USA 2007, 104, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.-M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative Repair of Oxidized Bases in the Human Genome Is Mediated by NEIL1 DNA Glycosylase Together with Replication Proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berquist, B.R.; Wilson, D.M. Pathways for Repairing and Tolerating the Spectrum of Oxidative DNA Lesions. Cancer Lett. 2012, 327, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bj Rås, K.Ø.; Sousa, M.M.L.; Sharma, A.; Fonseca, D.M.; S Gaard, C.K.; Bj Rås, M.; Otterlei, M. Monitoring of the Spatial and Temporal Dynamics of BER/SSBR Pathway Proteins, Including MYH, UNG2, MPG, NTH1 and NEIL1–3, during DNA Replication. Nucleic Acids Res. 2017, 45, 8291–8301. [Google Scholar] [CrossRef] [Green Version]
- Visnes, T.; Benítez-Buelga, C.; Cázares-Körner, A.; Sanjiv, K.; Hanna, B.M.F.; Mortusewicz, O.; Rajagopal, V.; Albers, J.J.; Hagey, D.W.; Bekkhus, T.; et al. Targeting OGG1 Arrests Cancer Cell Proliferation by Inducing Replication Stress. Nucleic Acids Res. 2020, 48, 12234–12251. [Google Scholar] [CrossRef]
- Boyer, A.-S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.-S. The Human Specialized DNA Polymerases and Non-B DNA: Vital Relationships to Preserve Genome Integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Gerhardt, J. Replication through Repetitive DNA Elements and Their Role in Human Diseases. In DNA Replication; Masai, H., Foiani, M., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; Volume 1042, pp. 549–581. ISBN 978-981-10-6954-3. [Google Scholar]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018, 174, 1127–1142.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shastri, N.; Tsai, Y.-C.; Hile, S.; Jordan, D.; Powell, B.; Chen, J.; Maloney, D.; Dose, M.; Lo, Y.; Anastassiadis, T.; et al. Genome-Wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol. Cell 2018, 72, 222–238.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Vasquez, K. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling Cell Type-Specific and Reprogrammable Human Replication Origin Signatures Associated with G-Quadruplex Consensus Motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef]
- Lansdorp, P.; Van Wietmarschen, N. Helicases FANCJ, RTEL1 and BLM Act on Guanine Quadruplex DNA in Vivo. Genes 2019, 10, 870. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shin-ya, K.; Niedzwiedz, W. FANCJ Couples Replication Past Natural Fork Barriers with Maintenance of Chromatin Structure. J. Cell Biol. 2013, 201, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Valton, A.-L.; Prioleau, M.-N. G-Quadruplexes in DNA Replication: A Problem or a Necessity? Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. Transcription–replication Conflicts: How They Occur and How They Are Resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.; Pasero, P. Interference between DNA Replication and Transcription as a Cause of Genomic Instability. Curr. Genom. 2012, 13, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.; Quinet, A.; Lin, Y.-L.; Davis, A.A.; Pasero, P.; Ayala, Y.M.; Vindigni, A. TDP-43 Dysfunction Results in R-Loop Accumulation and DNA Replication Defects. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Nardini, E.; Pellicioli, A.; Liberi, G. The Dark Side of RNA: DNA Hybrids. Mutat. Res. Mutat. Res. 2020, 784, 108300. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hua, Y.; Wang, J.; Li, L.; Yuan, J.; Zhang, B.; Wang, Z.; Ji, J.; Kong, D. RNA Polymerase III Is Required for the Repair of DNA Double-Strand Breaks by Homologous Recombination. Cell 2021, 184, 1314–1329.e10. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I Suppresses Genomic Instability by Preventing Interference between Replication and Transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Maffia, A.; Ranise, C.; Sabbioneda, S. From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. Int. J. Mol. Sci. 2020, 21, 1506. [Google Scholar] [CrossRef] [Green Version]
- Puget, N.; Miller, K.M.; Legube, G. Non-Canonical DNA/RNA Structures during Transcription-Coupled Double-Strand Break Repair: Roadblocks or Bona Fide Repair Intermediates? DNA Repair 2019, 81, 102661. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [Green Version]
- Primo, L.M.F.; Teixeira, L.K. DNA Replication Stress: Oncogenes in the Spotlight. Genet. Mol. Biol. 2019, 43, e20190138. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Halazonetis, T.D. Intragenic Origins due to Short G1 Phases Underlie Oncogene-Induced DNA Replication Stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Graziano, S.; Johnston, R.; Deng, O.; Zhang, J.; Gonzalo, S. Vitamin D/Vitamin D Receptor Axis Regulates DNA Repair during Oncogene-Induced Senescence. Oncogene 2016, 35, 5362–5376. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in Understanding the Complex Mechanisms of DNA Interstrand Cross-Link Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi Anaemia Pathway: New Players and New Functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- García-de-Teresa, B.; Rodríguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 15288. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabosseau, P.; Buhagiar-Labarchède, G.; Onclercq-Delic, R.; Lambert, S.; Debatisse, M.; Brison, O.; Amor-Guéret, M. Pyrimidine Pool Imbalance Induced by BLM Helicase Deficiency Contributes to Genetic Instability in Bloom Syndrome. Nat. Commun. 2011, 2, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; d’Adda di Fagagna, F. Breaking News: High-Speed Race Ends in Arrest—How Oncogenes Induce Senescence. Trends Cell Biol. 2007, 17, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Blumenfeld, B.; Ben-Zimra, M.; Simon, I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. Int. J. Mol. Sci. 2017, 18, 1138. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA Damage through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.; Walter, J.C.; Cimprich, K.A. Functional Uncoupling of MCM Helicase and DNA Polymerase Activities Activates the ATR-Dependent Checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Cotta-Ramusino, C.; McDonald, E.R.; Hurov, K.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. A DNA Damage Response Screen Identifies RHINO, a 9-1-1 and TopBP1 Interacting Protein Required for ATR Signaling. Science 2011, 332, 1313–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey-Boltz, L.A.; Kemp, M.G.; Capp, C.; Sancar, A. RHINO Forms a Stoichiometric Complex with the 9-1-1 Checkpoint Clamp and Mediates ATR-Chk1 Signaling. Cell Cycle 2015, 14, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Duursma, A.M.; Driscoll, R.; Elias, J.E.; Cimprich, K.A. A Role for the MRN Complex in ATR Activation through TOPBP1 Recruitment. Mol. Cell 2013, 50, 116–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.; Cimprich, K.A. The Structural Determinants of Checkpoint Activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef] [Green Version]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 Acts at Stalled Replication Forks to Maintain Genome Integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR Kinase by the RPA-Binding Protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. CB 2016, 26, 3257–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D. Preventing Replication Fork Collapse to Maintain Genome Integrity. DNA Repair 2015, 32, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Fréon, K.; Lambert, S.A.E. The End-Joining Factor Ku Acts in the End-Resection of Double Strand Break-Free Arrested Replication Forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 Nucleases Degrade Reversed Forks and Elicit MUS81-Dependent Fork Rescue in BRCA2-Deficient Cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Billing, D.; Horiguchi, M.; Wu-Baer, F.; Taglialatela, A.; Leuzzi, G.; Alvarez Nanez, S.; Jiang, W.; Zha, S.; Szabolcs, M.; Lin, C.-S.; et al. The BRCT Domains of the BRCA1 and BARD1 Tumor Suppressors Differentially Regulate Homology-Directed Repair and Stalled Fork Protection. Mol. Cell 2018, 72, 127–139.e8. [Google Scholar] [CrossRef] [Green Version]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1-BARD1 Promotes Replication Fork Protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 Protects Nascent DNA from Mre11-Dependent Degradation and Promotes Continuous DNA Synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Zadorozhny, K.; Sannino, V.; Beláň, O.; Mlčoušková, J.; Špírek, M.; Costanzo, V.; Krejčí, L. Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell Rep. 2017, 21, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication Fork Reversal Triggers Fork Degradation in BRCA2-Defective Cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef]
- Mason, J.M.; Chan, Y.-L.; Weichselbaum, R.W.; Bishop, D.K. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat. Commun. 2019, 10, 4410. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.-F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [Green Version]
- Higgs, M.R.; Stewart, G.S. Protection or Resection: BOD1L as a Novel Replication Fork Protection Factor. Nucleus 2016, 7, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Saintigny, Y.; Delacote, F.; Vares, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of Homologous Recombination Induced by Replication Inhibition in Mammalian Cells. Embo J. 2001, 20, 3861–3870. [Google Scholar] [CrossRef]
- Mills, K.D.; Ferguson, D.O.; Essers, J.; Eckersdorff, M.; Kanaar, R.; Alt, F.W. Rad54 and DNA Ligase IV Cooperate to Maintain Mammalian Chromatid Stability. Genes Dev. 2004, 18, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- Couedel, C.; Mills, K.D.; Barchi, M.; Shen, L.; Olshen, A.; Johnson, R.D.; Nussenzweig, A.; Essers, J.; Kanaar, R.; Li, G.C.; et al. Collaboration of Homologous Recombination and Nonhomologous End-Joining Factors for the Survival and Integrity of Mice and Cells. Genes Dev. 2004, 18, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.A.T.M.; Ray Chaudhuri, A. RIF1 Promotes Replication Fork Protection and Efficient Restart to Maintain Genome Stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Chen, B.-R.; Quinet, A.; Byrum, A.K.; Jackson, J.; Berti, M.; Thangavel, S.; Bredemeyer, A.L.; Hindi, I.; Mosammaparast, N.; Tyler, J.K.; et al. XLF and H2AX Function in Series to Promote Replication Fork Stability. J. Cell Biol. 2019, 218, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, V.; Robertson, K.; Bibikova, M.; Kim, E.; Grieco, D.; Gottesman, M.; Carroll, D.; Gautier, J. Mre11 Protein Complex Prevents Double-Strand Break Accumulation during Chromosomal DNA Replication. Mol. Cell 2001, 8, 137–147. [Google Scholar] [CrossRef]
- Costanzo, V. Brca2, Rad51 and Mre11: Performing Balancing Acts on Replication Forks. DNA Repair 2011, 10, 1060–1065. [Google Scholar] [CrossRef]
- Chaudhury, I.; Stroik, D.R.; Sobeck, A. FANCD2-Controlled Chromatin Access of the Fanconi-Associated Nuclease FAN1 Is Crucial for the Recovery of Stalled Replication Forks. Mol. Cell. Biol. 2014, 34, 3939–3954. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.E.; Lee, E.H.; Hendrickson, E.A.; Sobeck, A. CtIP Mediates Replication Fork Recovery in a FANCD2-Regulated Manner. Hum. Mol. Genet. 2014, 23, 3695–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Zhou, M.; Chai, Q.; Parrish, J.; Xue, D.; Patrick, S.M.; Turchi, J.J.; Yannone, S.M.; Chen, D.; Shen, B. Novel Function of the Flap Endonuclease 1 Complex in Processing Stalled DNA Replication Forks. EMBO Rep. 2005, 6, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehé, P.-M.; Gaillard, P.-H.L. Control of Structure-Specific Endonucleases to Maintain Genome Stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.-H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 Complex Is a SUMO E3 Ligase That Impacts on Replication Stress Outcome and Genome Stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, R.L.; Patel, S.; Rivard, R.S.; Smith, K.; Peters, A.A.; Bielinsky, A.-K.; Brown, E.J. RNF4 and PLK1 Are Required for Replication Fork Collapse in ATR-Deficient Cells. Genes Dev. 2013, 27, 2259–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malacaria, E.; Franchitto, A.; Pichierri, P. SLX4 Prevents GEN1-Dependent DSBs during DNA Replication Arrest Under Pathological Conditions in Human Cells. Sci. Rep. 2017, 7, 44464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The Structure-Specific Endonuclease Mus81 Contributes to Replication Restart by Generating Double-Strand DNA Breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef]
- Shimura, T.; Torres, M.J.; Martin, M.M.; Rao, V.A.; Pommier, Y.; Katsura, M.; Miyagawa, K.; Aladjem, M.I. Bloom’s Syndrome Helicase and Mus81 Are Required to Induce Transient Double-Strand DNA Breaks in Response to DNA Replication Stress. J. Mol. Biol. 2008, 375, 1152–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regairaz, M.; Zhang, Y.-W.; Fu, H.; Agama, K.K.; Tata, N.; Agrawal, S.; Aladjem, M.I.; Pommier, Y. Mus81-Mediated DNA Cleavage Resolves Replication Forks Stalled by Topoisomerase I-DNA Complexes. J. Cell Biol. 2011, 195, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 Promotes Common Fragile Site Expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; West, S.C. MUS81-EME2 Promotes Replication Fork Restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR Phosphorylates SMARCAL1 to Prevent Replication Fork Collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugger, K.; Chu, W.K.; Haahr, P.; Kousholt, A.N.; Beck, H.; Payne, M.J.; Hanada, K.; Hickson, I.D.; Sørensen, C.S. FBH1 Co-Operates with MUS81 in Inducing DNA Double-Strand Breaks and Cell Death Following Replication Stress. Nat. Commun. 2013, 4, 1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Otterlei, M.; Sommers, J.A.; Driscoll, H.C.; Dianov, G.L.; Kao, H.-I.; Bambara, R.A.; Brosh, R.M. WRN Helicase and FEN-1 Form a Complex upon Replication Arrest and Together Process Branchmigrating DNA Structures Associated with the Replication Fork. Mol. Biol. Cell 2004, 15, 734–750. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sommers, J.A.; Gary, R.K.; Friedrich-Heineken, E.; Hübscher, U.; Brosh, R.M. The Interaction Site of Flap Endonuclease-1 with WRN Helicase Suggests a Coordination of WRN and PCNA. Nucleic Acids Res. 2005, 33, 6769–6781. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Sengupta, S.; Yang, Q.; Linke, S.P.; Yanaihara, N.; Bradsher, J.; Blais, V.; McGowan, C.H.; Harris, C.C. BLM Helicase Facilitates Mus81 Endonuclease Activity in Human Cells. Cancer Res. 2005, 65, 2526–2531. [Google Scholar] [CrossRef] [Green Version]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 Drives Processing and Restart of Reversed Replication Forks in Human Cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, H.; Arter, M.; Gloggnitzer, J.; Teloni, F.; Wild, P.; Blanco, M.G.; Altmeyer, M.; Matos, J. A Mechanism for Controlled Breakage of Under-Replicated Chromosomes during Mitosis. Dev. Cell 2016, 39, 740–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To Skip or Not to Skip: Choosing Repriming to Tolerate DNA Damage. Mol. Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Sale Y-Family DNA Polymerases and Their Role in Tolerance of Cellular DNA Damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA Polymerases in Eukaryotes: What Makes Them Tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. Short Article PrimPol Bypasses UV Photoproducts during Eukaryotic Chromosomal DNA Replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef] [Green Version]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2020, 77, 461–474.e9. [Google Scholar] [CrossRef]
- Bunting, S.; Callen, E.; Wong, N.; Chen, H.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.-M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 Functions Independently of Homologous Recombination in DNA Interstrand Cross-Link Repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.; Steinberg-Shemer, O.; Unal, S.; Casadei, S.; Walsh, T.; Gumruk, F.; Shalev, S.; Shimamura, A.; Akarsu, N.A.; Tamary, H.; et al. Mechanism for Survival of Homozygous Nonsense Mutations in the Tumor Suppressor Gene BRCA1. Proc. Natl. Acad. Sci. USA 2018, 115, 5241–5246. [Google Scholar] [CrossRef] [Green Version]
- Venkitaraman, A.R. Tumour Suppressor Mechanisms in the Control of Chromosome Stability: Insights from BRCA2. Mol. Cells 2014, 37, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelot, C.; Guirouilh-Barbat, J.; Le Guen, T.; Dardillac, E.; Chailleux, C.; Canitrot, Y.; Lopez, B.S. The Cohesin Complex Prevents the End Joining of Distant DNA Double-Strand Ends. Mol. Cell 2016, 61, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelot, C.; Guirouilh-Barbat, J.; Lopez, B.S. The Cohesin Complex Prevents the End-Joining of Distant DNA Double-Strand Ends in S Phase: Consequences on Genome Stability Maintenance. Nucl. Austin Tex. 2016, 7, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leman, A.R.; Noguchi, C.; Lee, C.Y.; Noguchi, E. Human Timeless and Tipin Stabilize Replication Forks and Facilitate Sister-Chromatid Cohesion. J. Cell Sci. 2010, 123, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leman, A.R.; Noguchi, E. Local and Global Functions of Timeless and Tipin in Replication Fork Protection. Cell Cycle 2012, 11, 3945–3955. [Google Scholar] [CrossRef] [Green Version]
- Setiaputra, D.; Durocher, D. Shieldin—The Protector of DNA Ends. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Lee, D.-H.; Acharya, S.S.; Kwon, M.; Drane, P.; Guan, Y.; Adelmant, G.; Kalev, P.; Shah, J.; Pellman, D.; Marto, J.A.; et al. Dephosphorylation Enables the Recruitment of 53BP1 to Double-Strand DNA Breaks. Mol. Cell 2014, 54, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.-F.; Acharya, S.S.; Choe, K.N.; Nikhil, K.; Adelmant, G.; Satapathy, S.R.; Sharma, S.; Viccaro, K.; Rana, S.; Natarajan, A.; et al. A Mitotic CDK5-PP4 Phospho-Signaling Cascade Primes 53BP1 for DNA Repair in G1. Nat. Commun. 2019, 10, 4252. [Google Scholar] [CrossRef]
- Orthwein, A.; Fradet-Turcotte, A.; Noordermeer, S.; Canny, M.; Brun, C.; Strecker, J.; Escribano-Diaz, C.; Durocher, D. Mitosis Inhibits DNA Double-Strand Break Repair to Guard against Telomere Fusions. Science 2014, 344, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing Nonhomologous End Joining Suppresses DNA Repair Defects of Fanconi Anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 Corrupts DNA Repair in the Absence of the Fanconi Anemia Pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ning, S.; Wei, Z.; Xu, R.; Xu, X.; Xing, M.; Guo, R.; Xu, D. 53BP1 and BRCA1 Control Pathway Choice for Stalled Replication Restart. eLife 2017, 6. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.; Lee, A.; Wu, X. Cell Cycle-Dependent Complex Formation of BRCA1.CtIP.MRN Is Important for DNA Double-Strand Break Repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, P.; Jackson, S. Human CtIP Mediates Cell Cycle Control of DNA End Resection and Double Strand Break Repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and Annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Lambert, S.; Joubert, C.; Lopez, B.S. Overexpression of Mammalian Rad51 Does Not Stimulate Tumorigenesis While a Dominant-Negative Rad51 Affects Centrosome Fragmentation, Ploidy and Stimulates Tumorigenesis, in p53-Defective CHO Cells. Oncogene 2003, 22, 7587–7592. [Google Scholar] [CrossRef] [Green Version]
- Daboussi, F.; Thacker, J.; Lopez, B. Genetic Interactions between RAD51 and Its Paralogues for Centrosome Fragmentation and Ploidy Control, Independently of the Sensitivity to Genotoxic Stresses. Oncogene 2005, 24, 3691–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, C.S.; Simpson, P.J.; Wilson, C.R.; Thacker, J. Mammalian Recombination-Repair Genes XRCC2 and XRCC3 Promote Correct Chromosome Segregation [In Process Citation]. Nat. Cell Biol. 2000, 2, 757–761. [Google Scholar] [CrossRef]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Techer, H.; Debatisse, M.; Lopez, B. Spontaneous Slow Replication Fork Progression Elicits Mitosis Alterations in Homologous Recombination-Deficient Mammalian Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Olziersky, A.-M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild Replication Stress Causes Chromosome Mis-Segregation via Premature Centriole Disengagement. Nat. Commun. 2019, 10, 3585. [Google Scholar] [CrossRef] [Green Version]
- Bergoglio, V.; Boyer, A.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.; et al. DNA Synthesis by Pol Eta Promotes Fragile Site Stability by Preventing under-Replicated DNA in Mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81–EME1 Promote Sister Chromatid Separation by Processing Late Replication Intermediates at Common Fragile Sites during Mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication Stress Activates DNA Repair Synthesis in Mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, T.; Said, M.; Naim, V. DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes 2020, 11, 642. [Google Scholar] [CrossRef]
- Gasser, S.; Zhang, W.Y.L.; Tan, N.Y.J.; Tripathi, S.; Suter, M.A.; Chew, Z.H.; Khatoo, M.; Ngeow, J.; Cheung, F.S.G. Sensing of Dangerous DNA. Mech. Ageing Dev. 2017, 165, 33–46. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic Sensing of Immuno-Stimulatory DNA, the Enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Vilares, M.; Logarinho, E. Chromosomal Instability and pro-Inflammatory Response in Aging. Mech. Ageing Dev. 2019, 182, 111118. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS–STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Vashi, N.; Bakhoum, S.F. The Evolution of STING Signaling and Its Involvement in Cancer. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hartlova, A.; Erttmann, S.; Raffi, F.; Schmalz, A.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.; Kroger, A.; Nilsson, J.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A Prosurvival DNA Damage-Induced Cytoplasmic Interferon Response Is Mediated by End Resection Factors and Is Limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling by S-Phase-Specific DNA Damage in Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 109. [Google Scholar] [CrossRef] [Green Version]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 Deficiency Instigates cGAS-Mediated Inflammatory Signaling and Confers Sensitivity to Tumor Necrosis Factor-α-Mediated Cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.; Zhang, W.; Tan, N.; Khatoo, M.; Suter, M.; Tripathi, S.; Cheung, F.; Lim, W.; Tan, P.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 Acts at Stalled Replication Forks to Prevent Interferon Induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS Is Essential for Cellular Senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of Cytoplasmic DNases Is Implicated in Cytoplasmic DNA Accumulation and SASP in Senescent Cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Glück, S.; Ablasser, A. Innate Immunosensing of DNA in Cellular Senescence. Curr. Opin. Immunol. 2019, 56, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular Senescence and Senescence-Associated Secretory Phenotype via the cGAS-STING Signaling Pathway in Cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Moir, R.D.; Montag-Lowy, M.; Goldman, R.D. Dynamic Properties of Nuclear Lamins: Lamin B Is Associated with Sites of DNA Replication. J. Cell Biol. 1994, 125, 1201–1212. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.; Solovei, I.; Goldman, A.; Adam, S.; Shumaker, D.; Kinjo, M.; Cremer, T.; et al. The A- and B-Type Nuclear Lamin Networks: Microdomains Involved in Chromatin Organization and Transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [Green Version]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the Nuclear Interior—Life Outside the Lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear Lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Rusiñol, A.E.; Sinensky, M.S. Farnesylated Lamins, Progeroid Syndromes and Farnesyl Transferase Inhibitors. J. Cell Sci. 2006, 119, 3265–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, K.H.; Kennedy, B.K. When Lamins Go Bad: Nuclear Structure and Disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J. Nuclear Lamins and Laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. [Google Scholar] [CrossRef]
- Brull, A.; Morales Rodriguez, B.; Bonne, G.; Muchir, A.; Bertrand, A.T. The Pathogenesis and Therapies of Striated Muscle Laminopathies. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.; Munnich, A.; Le Merrer, M.; et al. Lamin a Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.; Gordon, L.; Glynn, M.; Singer, J.; Scott, L.; Erdos, M.; Robbins, C.; Moses, T.; Berglund, P.; et al. Recurrent de Novo Point Mutations in Lamin A Cause Hutchinson-Gilford Progeria Syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.; Gordon, L.; Collins, F.; Djabali, K. The Mutant Form of Lamin A That Causes Hutchinson-Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragnauth, C.; Warren, D.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C. Prelamin A Acts to Accelerate Smooth Muscle Cell Senescence and Is a Novel Biomarker of Human Vascular Aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation with the Vascular Pathology of Aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.-W.; Kriks, S.; et al. Human iPSC-Based Modeling of Late-Onset Disease via Progerin-Induced Aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Padiath, Q.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptacek, L.; Fu, Y. Lamin B1 Duplications Cause Autosomal Dominant Leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Padiath, Q.S. Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina. Front. Cell Dev. Biol. 2019, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Heng, M.Y.; Lin, S.-T.; Verret, L.; Huang, Y.; Kamiya, S.; Padiath, Q.S.; Tong, Y.; Palop, J.J.; Huang, E.J.; Ptáček, L.J.; et al. Lamin B1 Mediates Cell-Autonomous Neuropathology in a Leukodystrophy Mouse Model. J. Clin. Investig. 2013, 123, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Rolyan, H.; Tyurina, Y.Y.; Hernandez, M.; Amoscato, A.A.; Sparvero, L.J.; Nmezi, B.C.; Lu, Y.; Estécio, M.R.H.; Lin, K.; Chen, J.; et al. Defects of Lipid Synthesis Are Linked to the Age-Dependent Demyelination Caused by Lamin B1 Overexpression. J. Neurosci. J. Soc. Neurosci. 2015, 35, 12002–12017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative Stress Induces an ATM-Independent Senescence Pathway through p38 MAPK-Mediated Lamin B1 Accumulation. EMBO J. 2012, 31, 1080–1094. [Google Scholar] [CrossRef] [Green Version]
- Donadille, B.; D’Anella, P.; Auclair, M.; Uhrhammer, N.; Sorel, M.; Grigorescu, R.; Ouzounian, S.; Cambonie, G.; Boulot, P.; Laforet, P.; et al. Partial Lipodystrophy with Severe Insulin Resistance and Adult Progeria Werner Syndrome. Orphanet J. Rare Dis. 2013, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.A.; Martin, C.-A.; Greene, P.; Marsh, J.A.; Genomics England Research Consortium; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; et al. Heterozygous Lamin B1 and Lamin B2 Variants Cause Primary Microcephaly and Define a Novel Laminopathy. Genet. Med. 2020. [Google Scholar] [CrossRef]
- Cristofoli, F.; Moss, T.; Moore, H.W.; Devriendt, K.; Flanagan-Steet, H.; May, M.; Jones, J.; Roelens, F.; Fons, C.; Fernandez, A.; et al. De Novo Variants in LMNB1 Cause Pronounced Syndromic Microcephaly and Disruption of Nuclear Envelope Integrity. Am. J. Hum. Genet. 2020, 107, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Cao, H.; Liu, D.M.; Costain, G.A.; Charlton-Menys, V.; Rodger, N.W.; Durrington, P.N. Sequencing of the Reannotated LMNB2 Gene Reveals Novel Mutations in Patients with Acquired Partial Lipodystrophy. Am. J. Hum. Genet. 2006, 79, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, J.A.; Afawi, Z.; Bahlo, M.; Mauermann, M.; Misk, A.; Arsov, T.; Oliver, K.L.; Dahl, H.-H.M.; Shearer, A.E.; Smith, R.J.H.; et al. Mutation of the Nuclear Lamin Gene LMNB2 in Progressive Myoclonus Epilepsy with Early Ataxia. Hum. Mol. Genet. 2015, 24, 4483–4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. CB 2016, 26, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C. B-Type Lamins and Their Elusive Roles in Metazoan Cell Proliferation and Senescence. EMBO J. 2012, 31, 1058–1059. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Bourdeau, V.; Vernier, M.; Dabauvalle, M.-C.; Ferbeyre, G. Retinoblastoma-Independent Regulation of Cell Proliferation and Senescence by the p53–p21 Axis in Lamin A/C-Depleted Cells. Aging Cell 2011, 10, 789–797. [Google Scholar] [CrossRef]
- Huang, S.; Risques, R.; Martin, G.; Rabinovitch, P.; Oshima, J. Accelerated Telomere Shortening and Replicative Senescence in Human Fibroblasts Overexpressing Mutant and Wild-Type Lamin A. Exp. Cell Res. 2008, 314, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Candelario, J.; Sudhakar, S.; Navarro, S.; Reddy, S.; Comai, L. Perturbation of Wild-Type Lamin A Metabolism Results in a Progeroid Phenotype. Aging Cell 2008, 7, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of Mutant Lamin A Causes Progressive Changes in Nuclear Architecture in Hutchinson-Gilford Progeria Syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- Varela, I.; Cadinanos, J.; Pendas, A.M.; Gutierrez-Fernandez, A.; Folgueras, A.R.; Sanchez, L.M.; Zhou, Z.; Rodriguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated Ageing in Mice Deficient in Zmpste24 Protease Is Linked to p53 Signalling Activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Barcena, C.; Soria-Valles, C.; Ramsay, A.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.; Lopez-Otin, C. Nuclear Lamina Defects Cause ATM-Dependent NF-kappaB Activation and Link Accelerated Aging to a Systemic Inflammatory Response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Brassard, J.A.; Fekete, N.; Garnier, A.; Hoesli, C.A. Hutchinson–Gilford Progeria Syndrome as a Model for Vascular Aging. Biogerontology 2016, 17, 129–145. [Google Scholar] [CrossRef]
- Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Béréziat, V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells 2020, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.; Shanahan, C. Prelamin A Accelerates Vascular Calcification via Activation of the DNA Damage Response and Senescence-Associated Secretory Phenotype in Vascular Smooth Muscle Cells. Circ. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef] [Green Version]
- Vergnes, L.; Peterfy, M.; Bergo, M.; Young, S.; Reue, K. Lamin B1 Is Required for Mouse Development and Nuclear Integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but Not Lamin B1 Regulate Nuclear Mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Butin-Israeli, V.; Adam, S.; Hamanaka, R.; Goldman, A.; Lucas, C.; Shumaker, D.; Kosak, S.; Chandel, N.; Goldman, R. The Role of Nuclear Lamin B1 in Cell Proliferation and Senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Zaarour, N.; Bertrand, P. Oxydative Stress Alters Nuclear Shape through Lamins Dysregulation: A Route to Senescence. Nucleus 2012, 3, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.; Narita, M.; Perez-Mancera, P.; Bennett, D.; Chong, H.; et al. Redistribution of the Lamin B1 Genomic Binding Profile Affects Rearrangement of Heterochromatic Domains and SAHF Formation during Senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Donahue, G.; Otte, G.; Capell, B.; Nelson, D.; Cao, K.; Aggarwala, V.; Cruickshanks, H.; Rai, T.; McBryan, T.; et al. Lamin B1 Depletion in Senescent Cells Triggers Large-Scale Changes in Gene Expression and the Chromatin Landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Chojnowski, A.; Ong, P.; Zhao, T.; Common, J.; Lunny, D.; Lane, E.; Lee, S.; Vardy, L.; Stewart, C.; et al. Lamin B1 Fluctuations Have Differential Effects on Cellular Proliferation and Senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukášová, E.; Kovarˇík, A.; Bacˇíková, A.; Falk, M.; Kozubek, S. Loss of Lamin B Receptor Is Necessary to Induce Cellular Senescence. Biochem. J. 2017, 474, 281–300. [Google Scholar] [CrossRef]
- Wang, X.; Bi, X.; Yang, K.; Huang, Y.; Liu, Y.; Zhao, J. ROS/p38MAPK-Induced Lamin B1 Accumulation Promotes Chronic Kidney Disease-Associated Vascular Smooth Muscle Cells Senescence. Biochem. Biophys. Res. Commun. 2020, 531, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, G.; Marchio, C.; Gaetano, L.; Lupo, R.; Sapino, A. Pleomorphism of the Nuclear Envelope in Breast Cancer: A New Approach to an Old Problem. J. Cell Mol. Med. 2008, 12, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, G.; Maletta, F.; Asioli, S.; Annaratone, L.; Sapino, A.; Marchiò, C. “To Be or Not to Be in a Good Shape”: Diagnostic and Clinical Value of Nuclear Shape Irregularities in Thyroid and Breast Cancer. In Cancer Biology and the Nuclear Envelope; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014; pp. 101–121. ISBN 978-1-4899-8031-1. [Google Scholar]
- De Las Heras, J.I.; Schirmer, E.C. The Nuclear Envelope and Cancer: A Diagnostic Perspective and Historical Overview. Adv. Exp. Med. Biol. 2014, 773, 5–26. [Google Scholar] [CrossRef]
- Sengupta, D.; Mukhopadhyay, A.; Sengupta, K. Emerging Roles of Lamins and DNA Damage Repair Mechanisms in Ovarian Cancer. Biochem. Soc. Trans. 2020, 48, 2317–2333. [Google Scholar] [CrossRef]
- Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers 2020, 12, 3688. [Google Scholar] [CrossRef]
- Li, L.; Du, Y.; Kong, X.; Li, Z.; Jia, Z.; Cui, J.; Gao, J.; Wang, G.; Xie, K. Lamin B1 Is a Novel Therapeutic Target of Betulinic Acid in Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 4651–4661. [Google Scholar] [CrossRef] [Green Version]
- Izdebska, M.; Gagat, M.; Grzanka, A. Overexpression of Lamin B1 Induces Mitotic Catastrophe in Colon Cancer LoVo Cells and Is Associated with Worse Clinical Outcomes. Int. J. Oncol. 2018, 52, 89–102. [Google Scholar] [CrossRef]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 Is a Senescence-Associated Biomarker in Clear-Cell Renal Cell Carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef] [Green Version]
- Zy, Y.; Xy, J.; Rr, Z.; Cj, L.; Yx, R.; Zj, M.; Hl, Y.; Wg, S.; C, W.; Zy, J. Lamin B1 Deficiency Promotes Malignancy and Predicts Poor Prognosis in Gastric Cancer. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/32787434/ (accessed on 4 February 2021).
- Zhang, M.-Y.; Han, Y.-C.; Han, Q.; Liang, Y.; Luo, Y.; Wei, L.; Yan, T.; Yang, Y.; Liu, S.-L.; Wang, E.-H. Lamin B2 Promotes the Malignant Phenotype of Non-Small Cell Lung Cancer Cells by Upregulating Dimethylation of Histone 3 Lysine 9. Exp. Cell Res. 2020, 393, 112090. [Google Scholar] [CrossRef]
- Zhao, C.-C.; Chen, J.; Zhang, L.-Y.; Liu, H.; Zhang, C.-G.; Liu, Y. Lamin B2 Promotes the Progression of Triple Negative Breast Cancer via Mediating Cell Proliferation and Apoptosis. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiekema, M.; van Zandvoort, M.A.M.J.; Ramaekers, F.C.S.; Broers, J.L.V. Structural and Mechanical Aberrations of the Nuclear Lamina in Disease. Cells 2020, 9, 1884. [Google Scholar] [CrossRef]
- Shevelyov, Y.Y.; Ulianov, S.V. The Nuclear Lamina as an Organizer of Chromosome Architecture. Cells 2019, 8, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic Instability in Laminopathy-Based Premature Aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Reversal of the Cellular Phenotype in the Premature Aging Disease Hutchinson-Gilford Progeria Syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Seymour, I.; Markiewicz, E.; Bekker-Jensen, S.; Mailand, N.; Hutchison, C.J. Lamin A/C-Dependent Interaction with 53BP1 Promotes Cellular Responses to DNA Damage. Aging Cell 2015, 14, 162–169. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel Roles for A-Type Lamins in Telomere Biology and the DNA Damage Response Pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef] [PubMed]
- Mahen, R.; Hattori, H.; Lee, M.; Sharma, P.; Jeyasekharan, A.D.; Venkitaraman, A.R. A-Type Lamins Maintain the Positional Stability of DNA Damage Repair Foci in Mammalian Nuclei. PLoS ONE 2013, 8, e61893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant Nuclear Lamin A Leads to Progressive Alterations of Epigenetic Control in Premature Aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the Methyltransferase Suv39h1 Improves DNA Repair and Extends Lifespan in a Progeria Mouse Model. Nat. Commun. 2013, 4, 1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sun, L.; Wang, K.; Wu, D.; Trappio, M.; Witting, C.; Cao, K. Loss of H3K9me3 Correlates with ATM Activation and Histone H2AX Phosphorylation Deficiencies in Hutchinson-Gilford Progeria Syndrome. PLoS ONE 2016, 11, e0167454. [Google Scholar] [CrossRef]
- Ghosh, S.; Liu, B.; Wang, Y.; Hao, Q.; Zhou, Z. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep. 2015, 13, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Kugel, S.; Mostoslavsky, R. Chromatin and Beyond: The Multitasking Roles for SIRT6. Trends Biochem. Sci. 2014, 39, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The Sirtuin SIRT6 Regulates Lifespan in Male Mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Redwood, A.B.; Perkins, S.M.; Vanderwaal, R.P.; Feng, Z.; Biehl, K.J.; Gonzalez-Suarez, I.; Morgado-Palacin, L.; Shi, W.; Sage, J.; Roti-Roti, J.L.; et al. A Dual Role for A-Type Lamins in DNA Double-Strand Break Repair. Cell Cycle 2011, 10, 2549–2560. [Google Scholar] [CrossRef]
- di Masi, A.; D’Apice, M.; Ricordy, R.; Tanzarella, C.; Novelli, G. The R527H Mutation in LMNA Gene Causes an Increased Sensitivity to Ionizing Radiation. Cell Cycle 2008, 7, 2030–2037. [Google Scholar] [CrossRef] [Green Version]
- Mayca Pozo, F.; Tang, J.; Bonk, K.W.; Keri, R.A.; Yao, X.; Zhang, Y. Regulatory Cross-Talk Determines the Cellular Levels of 53BP1 Protein, a Critical Factor in DNA Repair. J. Biol. Chem. 2017, 292, 5992–6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, A.M.; Larrieu, D.; Warren, D.T.; Liu, Y.; Srivastava, S.; Smith, A.J.O.; Bowater, R.P.; Jackson, S.P.; Shanahan, C.M. Prelamin A Impairs 53BP1 Nuclear Entry by Mislocalizing NUP153 and Disrupting the Ran Gradient. Aging Cell 2016, 15, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Hunt, C.R.; Pandita, R.K.; Kumar, R.; Yang, C.-R.; Horikoshi, N.; Bachoo, R.; Serag, S.; Story, M.D.; Shay, J.W.; et al. Lamin A/C Depletion Enhances DNA Damage-Induced Stalled Replication Fork Arrest. Mol. Cell. Biol. 2013, 33, 1210–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, G.; Huang, M. Ribonucleotide Reductase Metallocofactor: Assembly, Maintenance and Inhibition. Front. Biol. 2014, 9, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butin-Israeli, V.; Adam, S.A.; Jain, N.; Otte, G.L.; Neems, D.; Wiesmüller, L.; Berger, S.L.; Goldman, R.D. Role of Lamin B1 in Chromatin Instability. Mol. Cell. Biol. 2015, 35, 884–898. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Sun, J.; Kono, K.; Horikoshi, Y.; Ikura, T.; Tong, X.; Haraguchi, T.; Tashiro, S. Regulation of Homologous Recombinational Repair by Lamin B1 in Radiation-Induced DNA Damage. FASEB J. 2015, 29, 2514–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Keijzers, G.; Akbari, M.; Ezra, M.B.; Hall, A.; Morevati, M.; Scheibye-Knudsen, M.; Gonzalo, S.; Bartek, J.; Bohr, V.A. Lamin A/C Promotes DNA Base Excision Repair. Nucleic Acids Res. 2019, gkz912. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Rusinol, A.E.; Sinensky, M.S.; Liu, J.; Shell, S.M.; Zou, Y. Involvement of Xeroderma Pigmentosum Group A (XPA) in Progeria Arising from Defective Maturation of Prelamin A. FASEB J. 2008, 22, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Regulation of Nucleotide Excision Repair by Nuclear Lamin b1. PLoS ONE 2013, 8, e69169. [Google Scholar] [CrossRef] [Green Version]
- Cesare, A.J.; Karlseder, J. A Three-State Model of Telomere Control over Human Proliferative Boundaries. Curr. Opin. Cell Biol. 2012, 24, 731–738. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The Protein Complex That Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Van Steensel, B.; Smogorzewska, A.; De Lange, T. TRF2 Protects Human Telomeres from End-to-End Fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; De Lange, T. p53- and ATM-Dependent Apoptosis Induced by Telomeres Lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Smogorzewska, A.; de Lange, T. Different Telomere Damage Signaling Pathways in Human and Mouse Cells. EMBO J. 2002, 21, 4338–4348. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Bilaud, T.; Brun, C.; Ancelin, K.; Koering, C.E.; Laroche, T.; Gilson, E. Telomeric Localization of TRF2, a Novel Human Telobox Protein. Nat. Genet. 1997, 17, 236–239. [Google Scholar] [CrossRef]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human Telomeres Contain Two Distinct Myb–related Proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-Loop Assembly in Vitro Involves Binding of TRF2 near the 3′ Telomeric Overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef] [Green Version]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-Resolution Fluorescence Imaging of Telomeres Reveals TRF2-Dependent T-Loop Formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.H.J.; de Lange, T. The Telomeric Protein TRF2 Binds the ATM Kinase and Can Inhibit the ATM-Dependent DNA Damage Response. PLoS Biol. 2004, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of Telomeres through Independent Control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.; Yates, J., 3rd; Denchi, E. A Two-Step Mechanism for TRF2-Mediated Chromosome-End Protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef] [Green Version]
- Feuerhahn, S.; Chen, L.; Luke, B.; Porro, A. No DDRama at Chromosome Ends: TRF2 Takes Centre Stage. Trends Biochem. Sci. 2015, 40, 275–285. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Shibuya, H.; Watanabe, Y. The Meiosis-Specific Modification of Mammalian Telomeres. Cell Cycle Georget. Tex. 2014, 13, 2024–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabbe, L.; Cesare, A.; Kasuboski, J.; Fitzpatrick, J.; Karlseder, J. Human Telomeres Are Tethered to the Nuclear Envelope during Postmitotic Nuclear Assembly. Cell Rep. 2012, 2, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Souaid, C.; Boulay, J.; Guerineau, V.; Noordermeer, D.; Crabbe, L. MadID, a Versatile Approach to Map Protein-DNA Interactions, Highlights Telomere-Nuclear Envelope Contact Sites in Human Cells. Cell Rep. 2018, 25, 2891–2903.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoult, N.; Schluth-Bolard, C.; Letessier, A.; Drascovic, I.; Bouarich-Bourimi, R.; Campisi, J.; Kim, S.; Boussouar, A.; Ottaviani, A.; Magdinier, F.; et al. Replication Timing of Human Telomeres Is Chromosome Arm-Specific, Influenced by Subtelomeric Structures and Connected to Nuclear Localization. PLoS Genet. 2010, 6, e1000920. [Google Scholar] [CrossRef] [Green Version]
- de Lange, T. Human Telomeres Are Attached to the Nuclear Matrix. EMBO J. 1992, 11, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Raz, V.; Vermolen, B.J.; Garini, Y.; Onderwater, J.J.M.; Mommaas-Kienhuis, M.A.; Koster, A.J.; Young, I.T.; Tanke, H.; Dirks, R.W. The Nuclear Lamina Promotes Telomere Aggregation and Centromere Peripheral Localization during Senescence of Human Mesenchymal Stem Cells. J. Cell Sci. 2008, 121, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allsopp, R.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.; Futcher, A.; Greider, C.; Harley, C. Telomere Length Predicts Replicative Capacity of Human Fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–10118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, M.; Chavez, E.; Vulto, I.; Lansdorp, P. Telomere Length in Hutchinson-Gilford Progeria Syndrome. Mech. Ageing Dev. 2009, 130, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Lee, S.; Aaronson, S. Role of Progerin-Induced Telomere Dysfunction in HGPS Premature Cellular Senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguado, J.; Sola-Carvajal, A.; Cancila, V.; Revêchon, G.; Ong, P.F.; Jones-Weinert, C.W.; Wallén Arzt, E.; Lattanzi, G.; Dreesen, O.; Tripodo, C.; et al. Inhibition of DNA Damage Response at Telomeres Improves the Detrimental Phenotypes of Hutchinson-Gilford Progeria Syndrome. Nat. Commun. 2019, 10, 4990. [Google Scholar] [CrossRef]
- Wood, A.M.; Rendtlew Danielsen, J.M.; Lucas, C.A.; Rice, E.L.; Scalzo, D.; Shimi, T.; Goldman, R.D.; Smith, E.D.; Le Beau, M.M.; Kosak, S.T. TRF2 and Lamin A/C Interact to Facilitate the Functional Organization of Chromosome Ends. Nat. Commun. 2014, 5, 5467. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.; Campbell, K.H.; Ford, C.C.; Stick, R.; Hutchison, C.J. The Role of Lamin LIII in Nuclear Assembly and DNA Replication, in Cell-Free Extracts of Xenopus Eggs. J. Cell Sci. 1991, 98 Pt 3, 271–279. [Google Scholar]
- Ellis, D.J.; Jenkins, H.; Whitfield, W.G.; Hutchison, C.J. GST-Lamin Fusion Proteins Act as Dominant Negative Mutants in Xenopus Egg Extract and Reveal the Function of the Lamina in DNA Replication. J. Cell Sci. 1997, 110 Pt 20, 2507–2518. [Google Scholar]
- Spann, T.P.; Moir, R.D.; Goldman, A.E.; Stick, R.; Goldman, R.D. Disruption of Nuclear Lamin Organization Alters the Distribution of Replication Factors and Inhibits DNA Synthesis. J. Cell Biol. 1997, 136, 1201–1212. [Google Scholar] [CrossRef]
- Moir, R.D.; Spann, T.P.; Herrmann, H.; Goldman, R.D. Disruption of Nuclear Lamin Organization Blocks the Elongation Phase of DNA Replication. J. Cell Biol. 2000, 149, 1179–1192. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Barbie, D.A.; Classon, M.; Dyson, N.; Harlow, E. Nuclear Organization of DNA Replication in Primary Mammalian Cells. Genes Dev. 2000, 14, 2855–2868. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M.; Itkonen, H.; Hillukkala, T.; Liu, Z.; Nasheuer, H.-P.; Schaarschmidt, D.; Pospiech, H.; Syväoja, J.E. Segregation of Replicative DNA Polymerases during S Phase: DNA Polymerase Ε, but Not DNA Polymerases α/Δ, Are Associated with Lamins throughout S Phase in Human Cells. J. Biol. Chem. 2012, 287, 33327–33338. [Google Scholar] [CrossRef] [Green Version]
- Shumaker, D.K.; Solimando, L.; Sengupta, K.; Shimi, T.; Adam, S.A.; Grunwald, A.; Strelkov, S.V.; Aebi, U.; Cardoso, M.C.; Goldman, R.D. The Highly Conserved Nuclear Lamin Ig-Fold Binds to PCNA: Its Role in DNA Replication. J. Cell Biol. 2008, 181, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Cobb, A.M.; Murray, T.V.; Warren, D.T.; Liu, Y.; Shanahan, C.M. Disruption of PCNA-Lamins A/C Interactions by Prelamin A Induces DNA Replication Fork Stalling. Nucl. Austin Tex. 2016, 7, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic Identification of Pathological Lamin A Interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Voncken, J.W.; Demmers, J.; Calis, C.; van Almen, G.; Pinto, Y.; Misteli, T. Identification of Differential Protein Interactors of Lamin A and Progerin. Nucl. Austin Tex. 2010, 1, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, B.A.; Liu, J.; Cartwright, B.M.; Liu, Y.; Breitman, M.; Wang, Y.; Jones, R.; Tang, H.; Rusinol, A.; Musich, P.R.; et al. Progerin Sequestration of PCNA Promotes Replication Fork Collapse and Mislocalization of XPA in Laminopathy-Related Progeroid Syndromes. FASEB J. 2017, 31, 3882–3893. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, K.; Campuzano, D.; Ma, W.; Sheinis, M.; Ho, B.; Brown, G.W.; Benchimol, S. Progerin-Induced Replication Stress Facilitates Premature Senescence in Hutchinson-Gilford Progeria Syndrome. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.R.; Nitta, R.T.; Frock, R.L.; Mounkes, L.; Barbie, D.A.; Stewart, C.L.; Harlow, E.; Kennedy, B.K. A-Type Lamins Regulate Retinoblastoma Protein Function by Promoting Subnuclear Localization and Preventing Proteasomal Degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9677–9682. [Google Scholar] [CrossRef] [Green Version]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef] [Green Version]
- Camps, J.; Wangsa, D.; Falke, M.; Brown, M.; Case, C.M.; Erdos, M.R.; Ried, T. Loss of Lamin B1 Results in Prolongation of S Phase and Decondensation of Chromosome Territories. FASEB J. 2014, 28, 3423–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA Replication Timing in the 3D Genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737. [Google Scholar] [CrossRef]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing Newly Replicated DNA Reveals Widespread Plasticity in Human Replication Timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.M.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular Maps of the Reorganization of Genome-Nuclear Lamina Interactions during Differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Duriez, B.; Chilaka, S.; Bercher, J.-F.; Hercul, E.; Prioleau, M.-N. Replication Dynamics of Individual Loci in Single Living Cells Reveal Changes in the Degree of Replication Stochasticity through S Phase. Nucleic Acids Res. 2019, 47, 5155–5169. [Google Scholar] [CrossRef] [Green Version]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Desprat, R.; Trevilla-Garcia, C.; Cornacchia, D.; Schwerer, H.; Sasaki, T.; Sima, J.; Fells, T.; Studer, L.; Lemaitre, J.-M.; et al. DNA Replication Timing Alterations Identify Common Markers between Distinct Progeroid Diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E10972–E10980. [Google Scholar] [CrossRef] [Green Version]
- Li, B.X.; Chen, J.; Chao, B.; Zheng, Y.; Xiao, X. A Lamin-Binding Ligand Inhibits Homologous Recombination Repair of DNA Double-Strand Breaks. ACS Cent. Sci. 2018, 4, 1201–1210. [Google Scholar] [CrossRef]
- Stratigi, K.; Chatzidoukaki, O.; Garinis, G.A. DNA Damage-Induced Inflammation and Nuclear Architecture. Mech. Ageing Dev. 2017, 165, 17–26. [Google Scholar] [CrossRef]
- Kristiani, L.; Kim, M.; Kim, Y. Role of the Nuclear Lamina in Age-Associated Nuclear Reorganization and Inflammation. Cells 2020, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Tseng, C.; Hambright, W.S.; Matre, P.; Lin, C.-Y.; Chanda, P.; Chen, W.; Gu, J.; Ravuri, S.; Cui, Y.; et al. Cytoskeleton Stiffness Regulates Cellular Senescence and Innate Immune Response in Hutchinson-Gilford Progeria Syndrome. Aging Cell 2020. [Google Scholar] [CrossRef]
- Di Micco, A.; Frera, G.; Lugrin, J.; Jamilloux, Y.; Hsu, E.-T.; Tardivel, A.; De Gassart, A.; Zaffalon, L.; Bujisic, B.; Siegert, S.; et al. AIM2 Inflammasome Is Activated by Pharmacological Disruption of Nuclear Envelope Integrity. Proc. Natl. Acad. Sci. USA 2016, 113, E4671–E4680. [Google Scholar] [CrossRef] [Green Version]
- Ashapkin, V.V.; Kutueva, L.I.; Kurchashova, S.Y.; Kireev, I.I. Are There Common Mechanisms Between the Hutchinson-Gilford Progeria Syndrome and Natural Aging? Front. Genet. 2019, 10, 455. [Google Scholar] [CrossRef] [Green Version]
- Coll-Bonfill, N.; Cancado de Faria, R.; Bhoopatiraju, S.; Gonzalo, S. Calcitriol Prevents RAD51 Loss and cGAS-STING-IFN Response Triggered by Progerin. Proteomics 2019, e1800406. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA Breaks and Chromosome Pulverization from Errors in Mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohard, S.; Bourgeois, N.; Maillet, L.; Gautier, F.; Fétiveau, A.; Lasla, H.; Nguyen, F.; Vuillier, C.; Dumont, A.; Moreau-Aubry, A.; et al. STING-Dependent Paracriny Shapes Apoptotic Priming of Breast Tumors in Response to Anti-Mitotic Treatment. Nat. Commun. 2020, 11, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S. Chemical Inhibition of NAT10 Corrects Defects of Laminopathic Cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmus, G.; Larrieu, D.; Barros, A.C.; Collins, C.; Abrudan, M.; Demir, M.; Geisler, N.J.; Lelliott, C.J.; White, J.K.; Karp, N.A.; et al. Targeting of NAT10 Enhances Healthspan in a Mouse Model of Human Accelerated Aging Syndrome. Nat. Commun. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yao, M.; Wu, Y.; Ma, N.; Liu, H.; Zhang, B. N-Acetyltransferase 10 Promotes Micronuclei Formation to Activate the Senescence-Associated Secretory Phenotype Machinery in Colorectal Cancer Cells. Transl. Oncol. 2020, 13, 100783. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.; Tutt, A.; Johnson, D.; Richardson, T.; Santarosa, M.; Dillon, K.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: The First Synthetic Lethal Targeted Therapy. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Peng, H. STING-Cytosolic DNA Sensing: The Backbone for an Effective Tumor Radiation Therapy. Ann. Transl. Med. 2016, 4, 60. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS Is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an Important Pathway in Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.; Cottu, P.; Sastre-Garau, X.; et al. miR-141 and miR-200a Act on Ovarian Tumorigenesis by Controlling Oxidative Stress Response. Nat. Med. 2011, 17, 1627–1635. [Google Scholar] [CrossRef]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The Role of Reactive Oxygen Species and Metabolism on Cancer Cells and Their Microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.-R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Duménil, A.-M.; Manel, N.; et al. ESCRT III Repairs Nuclear Envelope Ruptures during Cell Migration to Limit DNA Damage and Cell Death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Laberge, R.; Awad, P.; Campisi, J.; Desprez, P. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in Micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear Envelope Assembly Defects Link Mitotic Errors to Chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS Drives Non-Canonical Inflammasome Activation in Age-Related Macular Degeneration. Nat. Med. 2018, 24, 50–61. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willaume, S.; Rass, E.; Fontanilla-Ramirez, P.; Moussa, A.; Wanschoor, P.; Bertrand, P. A Link between Replicative Stress, Lamin Proteins, and Inflammation. Genes 2021, 12, 552. https://doi.org/10.3390/genes12040552
Willaume S, Rass E, Fontanilla-Ramirez P, Moussa A, Wanschoor P, Bertrand P. A Link between Replicative Stress, Lamin Proteins, and Inflammation. Genes. 2021; 12(4):552. https://doi.org/10.3390/genes12040552
Chicago/Turabian StyleWillaume, Simon, Emilie Rass, Paula Fontanilla-Ramirez, Angela Moussa, Paul Wanschoor, and Pascale Bertrand. 2021. "A Link between Replicative Stress, Lamin Proteins, and Inflammation" Genes 12, no. 4: 552. https://doi.org/10.3390/genes12040552
APA StyleWillaume, S., Rass, E., Fontanilla-Ramirez, P., Moussa, A., Wanschoor, P., & Bertrand, P. (2021). A Link between Replicative Stress, Lamin Proteins, and Inflammation. Genes, 12(4), 552. https://doi.org/10.3390/genes12040552