
Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine


Abstract
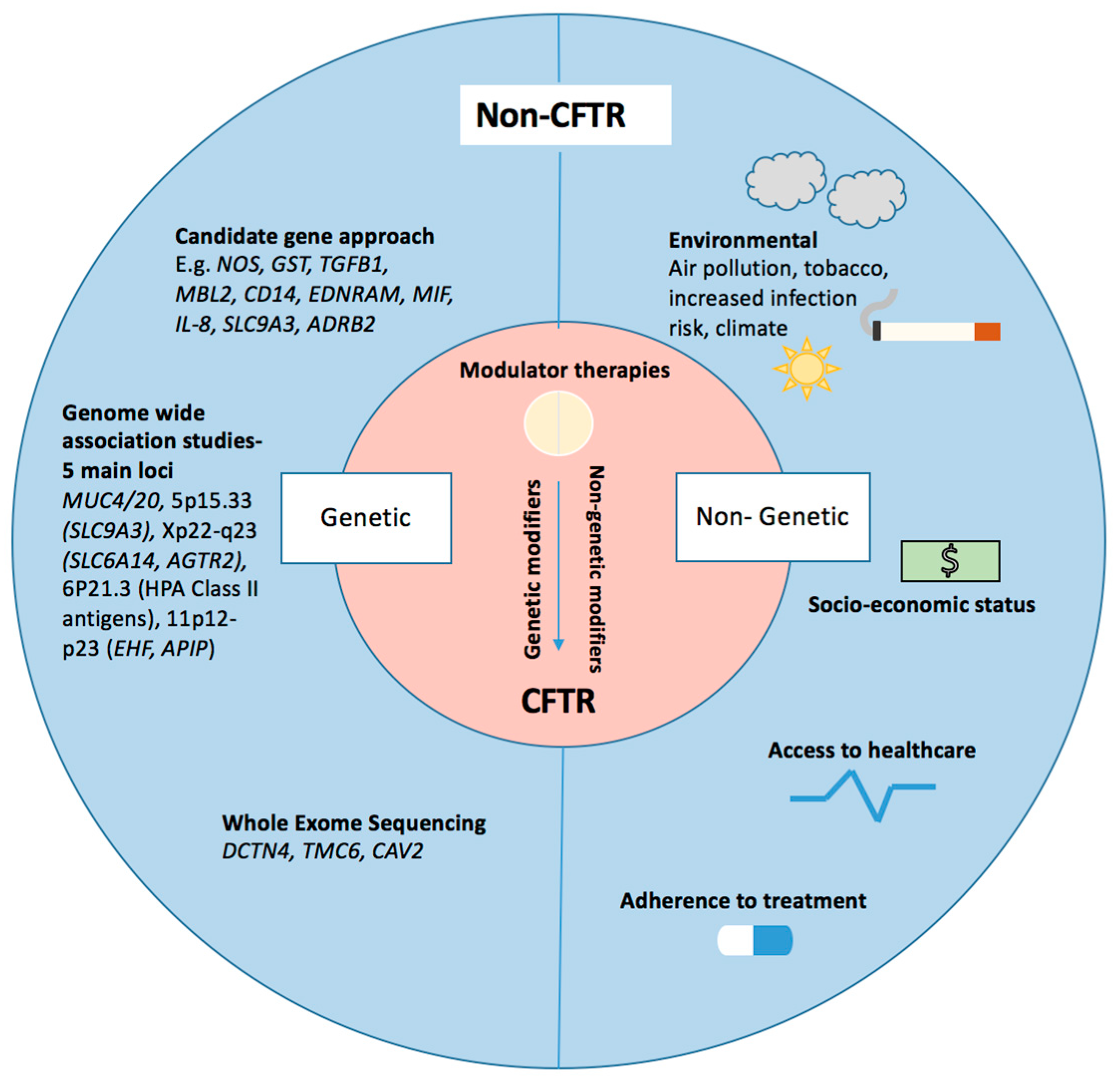
:1. Introduction
2. Non-CFTR Disease Modifiers
3. Genetic Modifiers of CF Lung Disease
4. Candidate Gene Studies
4.1. Nitric Oxide Synthase (NOS)
4.2. Glutathione and Glutathione-S-Transferase (GST)
4.3. Transforming Growth Factor β 1 (TGFβ1)
4.4. Mannose Binding Lectin 2 (MBL2)
4.5. Cell Surface Receptors
4.6. Macrophage Migration Inhibitory Factor (MIF)
4.7. Cytokines
4.8. Ion Channels
4.9. Mucins
4.10. Genes Involved in Drug Responses
5. Genome-Wide Associations Studies (GWAS)
6. Whole Exome Sequencing Studies
7. Non-Genetic Modifiers of Lung Disease Severity
8. CFTR Modulator Therapies
9. Will Disease Modifiers Continue to Be Relevant in Patients Experiencing Restored CFTR Function from CFTR Modulators?
10. Is There a Role for Modifiers in the Response to Modulator Therapies?
11. Potential Non-Genetic Modifiers of Response to HEMT
12. Discussion
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redondo, M.; Keyt, H.; Dhar, R.; Chalmers, J.D. Global impact of bronchiectasis and cystic fibrosis. Breathe 2016, 12, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Klein, K.; Hartel, G.; Wainwright, C.E.; Bell, S.C.; Anderson, G.J.; Reid, D.W. Mutations in the HFE gene can be associated with increased lung disease severity in cystic fibrosis. Gene 2019, 683, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Viviani, L.; Zolin, A.; MacNeill, S.; Hatziagorou, E.; Ellemunter, H.; Drevinek, P.; Gulmans, V.; Krivec, U.; Olesen, H.; et al. Factors associated with FEV1 decline in cystic fibrosis: Analysis of the ECFS Patient Registry. Eur. Respir. J. 2013, 43, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Kerem, E.; Corey, M.; Kerem, B.S.; Rommens, J.; Markiewicz, D.; Levison, H.; Tsui, L.-C.; Durie, P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N. Engl. J. Med. 1990, 323, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- Santis, G.; Osborne, L.; Knight, R.; Hodson, M. Independent genetic determinants of pancreatic and pulmonary status in cystic fibrosis. Lancet 1990, 336, 1081–1084. [Google Scholar] [CrossRef]
- Vanscoy, L.L.; Blackman, S.M.; Collaco, J.M.; Bowers, A.; Lai, T.; Naughton, K.; Algire, M.; McWilliams, R.; Beck, S.; Hoover-Fong, J.; et al. Heritability of Lung Disease Severity in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekus, F.; Ballmann, M.; Bronsveld, I.; Bijman, J.; Veeze, H.; Tümmler, B. Categories of deltaF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res. 2000, 3, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M. Multiple roles of nitric oxide in the airways. Thorax 2003, 58, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Dötsch, J.; Puls, J.; Klimek, T.; Rascher, W. Reduction of neuronal and inducible nitric oxide synthase gene expression in patients with cystic fibrosis. Eur. Arch. Oto-Rhino-Laryngol. 2002, 259, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Grasemann, H.; Deykin, A.; Silverman, E.K.; Yandava, C.N.; Israel, E.; Wand, M.; Drazen, J.M. Exhaled nitric oxide in patients with asthma: Association with NOS1 genotype. Am. J. Respir. Crit. Care Med. 2000, 162, 2043–2047. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Knauer, N.; Büscher, R.; Hübner, K.; Drazen, J.M.; Ratjen, F. Airway Nitric Oxide Levels in Cystic Fibrosis Patients Are Related to a Polymorphism in the Neuronal Nitric Oxide Synthase Gene. Am. J. Respir. Crit. Care Med. 2000, 162, 2172–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasemann, H.; Gravesande, K.S.V.; Büscher, R.; Knauer, N.; Silverman, E.S.; Palmer, L.J.; Drazen, J.M.; Ratjen, F. Endothelial Nitric Oxide Synthase Variants in Cystic Fibrosis Lung Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 390–394. [Google Scholar] [CrossRef]
- Texereau, J.; Marullo, S.; Hubert, D.; Coste, J.; Dusser, D.J.; Dall’Ava-Santucci, J.; Dinh-Xuan, A.T. Nitric oxide synthase 1 as a potential modifier gene of decline in lung function in patients with cystic fibrosis. Thorax 2004, 59, 156–158. [Google Scholar] [CrossRef] [Green Version]
- Flamant, C.; Henrion-Caude, A.; Boëlle, P.-Y.; Brémont, F.; Brouard, J.; Delaisi, B.; Duhamel, J.-F.; Marguet, C.; Roussey, M.; Miesch, M.-C.; et al. Glutathione-S-transferase M1, M3, P1 and T1 polymorphisms and severity of lung disease in children with cystic fibrosis. Pharmacogenetics 2004, 14, 295–301. [Google Scholar] [CrossRef]
- Marson, F.A.d.L.; Bertuzzo, C.S.; Secolin, R.; Ribeiro, A.F.; Ribeiro, J.D. Genetic interaction of GSH metabolic pathway genes in cystic fibrosis. BMC Med. Genet. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, J.; Thomson, A.H. Contribution of genetic factors other than CFTR to disease severity in cystic fibrosis. Thorax 1998, 53, 1018–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic Modifiers of Lung Disease in Cystic Fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.L.; Clancy, J.P. TGFbeta as a therapeutic target in cystic fibrosis. Expert Opin. Ther. Targets 2018, 22, 177–189. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Laurie, S.; Super, M.; Pravica, V.; Schwarz, M.J.; Webb, A.K.; Hutchinson, I.V. TGF-beta 1 genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax 2000, 55, 459–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.; Jansen, S.; Tamm, S.; Wienker, T.F.; Tümmler, B.; Stanke, F. Transmission ratio distortion and maternal effects confound the analysis of modulators of cystic fibrosis disease severity on 19qeur. J. Hum. Genet. 2007, 15, 774–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillot, L.; Beucher, J.; Tabary, O.; Le Rouzic, P.; Clement, A.; Corvol, H. Lung disease modifier genes in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 83–93. [Google Scholar] [CrossRef]
- Collaco, J.M.; Vanscoy, L.; Bremer, L.; McDougal, K.; Blackman, S.M.; Bowers, A.; Naughton, B.K.; Jennings, J.; Ellen, J.; Cutting, G.R. Interactions Between Secondhand Smoke and Genes That Affect Cystic Fibrosis Lung Disease. JAMA 2008, 299, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M. The role of mannose-binding lectin in health and disease. Mol. Immunol. 2003, 40, 423–429. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Fleming, G.B.; Hill, A.T.; Kilpatrick, D.C. Impact of mannose-binding lectin insufficiency on the course of cystic fibrosis: A review and meta-analysis. Glycobiology 2010, 21, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfman, R.; Sandford, A.; Taylor, C.; Huang, B.; Frangolias, D.; Wang, Y.; Sang, R.; Pereira, L.; Sun, L.; Berthiaume, Y.; et al. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J. Clin. Investig. 2008, 118, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, M.; Sjöholm, A.G.; Eriksson, L.; Thiel, S.; Jensenius, J.C.; Segelmark, M.; Truedsson, L. Deficiency of the mannan-binding lectin pathway of complement and poor outcome in cystic fibrosis: Bacterial colonization may be decisive for a relationship. Clin. Exp. Immunol. 2005, 139, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, H.V.; Jensenius, J.C.; Steffensen, R.; Thiel, S.; Schiøtz, P.O. The mannan-binding lectin pathway and lung disease in cystic fibrosis--disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin. Immunol. 2006, 121, 324–331. [Google Scholar] [CrossRef]
- Stanke, F.; Becker, T.; Kumar, V.; Hedtfeld, S.; Becker, C.; Cuppens, H.; Tamm, S.; Yarden, J.; Laabs, U.; Siebert, B.; et al. Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia. J. Med. Genet. 2010, 48, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, D.S.; Allen, J.; Elrayess, M.; Fidler, K.; Klein, N.; Jaffe, A. Modifier effect of the Toll-like receptor 4 D299G polymorphism in children with cystic fibrosis. Arch. Immunol. Et Ther. Exp. 2006, 54, 271–276. [Google Scholar] [CrossRef]
- Park, J.E.; Yung, R.; Stefanowicz, D.; Shumansky, K.; Akhabir, L.; Durie, P.R.; Corey, M.; Zielenski, J.; Dorfman, R.I.; Daley, D.; et al. Cystic fibrosis modifier genes related to Pseudomonas aeruginosa infection. Genes Immun. 2011, 12, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Alexis, N.; Eldridge, M.; Reed, W.; Bromberg, P.; Peden, D.B. CD14-dependent airway neutrophil response to inhaled LPS: Role of atopy. J. Allergy Clin. Immunol. 2001, 107, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.; Laing, I.A.; Zhang, G.; Brennan, S.; Winfield, K.; Sly, P.; Stick, S.; Goldblatt, J.; LeSouef, P.N. CD14 C-159T and early infection with Pseudomonas aeruginosa in children with cystic fibrosis. Respir. Res. 2005, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darrah, R.; McKone, E.; O’Connor, C.; Rodgers, C.; Genatossio, A.; McNamara, S.; Gibson, R.; Elborn, J.S.; Ennis, M.; Gallagher, C.G.; et al. EDNRA variants associate with smooth muscle mRNA levels, cell proliferation rates, and cystic fibrosis pulmonary disease severity. Physiol. Genom. 2010, 41, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plant, B.J.; Gallagher, C.G.; Bucala, R.; Baugh, J.A.; Chappell, S.; Morgan, L.; O’Connor, C.M.; Morgan, K.; Donnelly, S.C. Cystic Fibrosis, Disease Severity, and a Macrophage Migration Inhibitory Factor Polymorphism. Am. J. Respir. Crit. Care Med. 2005, 172, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Melotti, P.; Mafficini, A.; Lebecque, P.; Ortombina, M.; Leal, T.; Pintani, E.; Pepermans, X.; Sorio, C.; Assael, B.M. Impact of MIF Gene Promoter Polymorphism on F508del Cystic Fibrosis Patients. PLoS ONE 2014, 9, e114274. [Google Scholar] [CrossRef] [Green Version]
- Hillian, A.; Londono, D.; Dunn, J.; Goddard, K.; Pace, R.; Knowles, M.; Drumm, M.; CF Gene Modifier Study Group. Modulation of cystic fibrosis lung disease by variants in interleukin-8. Genes Immun. 2008, 9, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvol, H.; Burchard, E.G. Pharmacogenetic response to albuterol among asthmatics. Pharmacogenomics 2008, 9, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Murphy, A.; Zou, F.; Gerard, C.; Klanderman, B.; Bs, B.S.; Lazarus, R.; ChristopherGarcia, K.E.d.M.; Celedón, J.C.; Drumm, M.; et al. IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 2009, 44, 580–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, F.; Hector, A.; Hedtfeld, S.; Hartl, D.; Griese, M.; Tümmler, B.; Mall, M.A. An informative intragenic microsatellite marker suggests the IL-1 receptor as a genetic modifier in cystic fibrosis. Eur. Respir. J. 2017, 50, 1700426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfman, R.; Taylor, C.; Lin, F.; Sun, L.; Sandford, A.; Paré, P.; Berthiaume, Y.; Corey, M.; Durie, P.; Zielenski, J.; et al. Modulatory effect of the SLC9A3 gene on susceptibility to infections and pulmonary function in children with cystic fibrosis. Pediatr. Pulmonol. 2010, 46, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.L.; Chalfant, M.L.; Jovov, B.; Lockhart, J.P.; Parker, S.B.; Fuller, C.M.; Stanton, B.A.; Benos, D.J. The cytosolic termini of the beta- and gamma-ENaC subunits are involved in the functional interactions between cystic fibrosis transmembrane conductance regulator and epithelial sodium channel. J. Biol. Chem. 2000, 275, 27947–27956. [Google Scholar] [CrossRef]
- Stanke, F.; Becker, T.; Cuppens, H.; Kumar, V.; Cassiman, J.-J.; Jansen, S.; Radojkovic, D.; Siebert, B.; Yarden, J.; Ussery, D.W.; et al. The TNFα receptor TNFRSF1A and genes encoding the amiloride-sensitive sodium channel ENaC as modulators in cystic fibrosis. Qual. Life Res. 2006, 119, 331–343. [Google Scholar] [CrossRef]
- Shei, R.-J.; Peabody, J.E.; Kaza, N.; Rowe, S.M. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol. 2018, 43, 152–165. [Google Scholar] [CrossRef]
- O’Riordan, T.G.; Donn, K.H.; Hodsman, P.; Ansede, J.H.; Newcomb, T.; Lewis, S.A.; Flitter, W.D.; White, V.S.; Johnson, M.R.; Montgomery, A.B.; et al. Acute Hyperkalemia Associated with Inhalation of a Potent ENaC Antagonist: Phase 1 Trial of GS-9411. J. Aerosol Med. Pulm. Drug Deliv. 2014, 27, 200–208. [Google Scholar] [CrossRef]
- Caldwell, R.A.; Boucher, R.C.; Stutts, M.J. Neutrophil elastase activates near-silent epithelial Na+channels and increases airway epithelial Na+transport. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L813–L819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niv, Y.; Ho, S.B.; Rokkas, T. Mucin secretion in cystic fibrosis—A systematic review. Dig. Dis. 2020. [Google Scholar] [CrossRef]
- Guo, X.; Pace, R.G.; Stonebraker, J.R.; Commander, C.W.; Dang, A.T.; Drumm, M.L.; Harris, A.; Zou, F.; Swallow, D.M.; Wright, F.A.; et al. Mucin variable number tandem repeat polymorphisms and severity of cystic fibrosis lung disease: Significant association with MUC5AC. PLoS ONE 2011, 6, e25452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.A.; Konstan, M.W.; Darrah, R.J.; Schluchter, M.D.; Xue, L.; Londono, U.; Goddard, K.A.; Drumm, M.L.; Storfer-Isser, A. Beta 2 adrenergic receptor polymorphisms in cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 544–550. [Google Scholar] [CrossRef] [PubMed]
- De Paiva, A.C.; Marson, F.A.; Ribeiro, J.D.; Bertuzzo, C.S. Asthma: Gln27Glu and Arg16Gly polymorphisms of the beta2-adrenergic receptor gene as risk factors. Allerg. Asthma Clin. Immunol. 2014, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvol, H.; Nathan, N.; Charlier, C.; Chadelat, K.; Le Rouzic, P.; Tabary, O.; Fauroux, B.; Henrion-Caude, A.; Feingold, J.; Boelle, P.-Y.; et al. Glucocorticoid receptor gene polymorphisms associated with progression of lung disease in young patients with cystic fibrosis. Respir. Res. 2007, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Corvol, H.; Blackman, S.M.; Boëlle, P.-Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6, 8382. [Google Scholar] [CrossRef] [Green Version]
- Wright, F.A.; Strug, L.J.; Doshi, V.K.; Commander, C.W.; Blackman, S.M.; Sun, L.; Berthiaume, Y.; Cutler, D.M.; Cojocaru, A.; Collaco, J.M.; et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13. Nat. Genet. 2011, 43, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Harley, I.T.W.; Henderson, L.B.; Aronow, B.J.; Vietor, I.; Huber, L.A.; Harley, J.B.; Kilpatrick, J.R.; Langefeld, C.D.; Williams, A.H.; et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nat. Cell Biol. 2009, 458, 1039–1042. [Google Scholar] [CrossRef]
- Song, D.; Cahn, D.; Duncan, G.A. Mucin Biopolymers and Their Barrier Function at Airway Surfaces. Langmuir 2020, 36, 12773–12783. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Soave, D.; Miller, M.R.; Keenan, K.; Lin, F.; Gong, J.; Chiang, T.; Stephenson, A.L.; Durie, P.; Rommens, J.; et al. Unraveling the complex genetic model for cystic fibrosis: Pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Qual. Life Res. 2013, 133, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Soave, D.; Corvol, H.; Panjwani, N.; Gong, J.; Li, W.; Boëlle, P.-Y.; Durie, P.R.; Paterson, A.D.; Rommens, J.M.; Strug, L.J.; et al. A Joint Location-Scale Test Improves Power to Detect Associated SNPs, Gene Sets, and Pathways. Am. J. Hum. Genet. 2015, 97, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Bradford, E.M.; Sartor, M.A.; Gawenis, L.R.; Clarke, L.L.; Shull, G.E. Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am. J. Physiol. Liver Physiol. 2009, 296, G886–G898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Rommens, J.M.; Corvol, H.; Li, W.; Li, X.; Chiang, T.A.; Lin, F.; Dorfman, R.; Busson, P.-F.; Parekh, R.V.; et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat. Genet. 2012, 44, 562–569. [Google Scholar] [CrossRef]
- Strug, L.J.; Gonska, T.; He, G.; Keenan, K.; Ip, W.; Boëlle, P.-Y.; Lin, F.; Panjwani, N.; Gong, J.; Li, W.; et al. Cystic fibrosis gene modifierSLC26A9modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 2016, 25, 4590–4600. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, M.; Park, A.J.; Ahmadi, S.; Roach, E.J.; Wu, Y.S.; Struder-Kypke, M.; Lam, S.J.; Bear, C.E.; Khursigara, C.M. SLC6A14 Is a Genetic Modifier of Cystic Fibrosis That Regulates Pseudomonas aeruginosa Attachment to Human Bronchial Epithelial Cells. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, S.; Wu, Y.-S.; Li, M.; Ip, W.; Lloyd-Kuzik, A.; Di Paola, M.; Du, K.; Xia, S.; Lew, A.; Bozoky, Z.; et al. Augmentation of Cystic Fibrosis Transmembrane Conductance Regulator Function in Human Bronchial Epithelial Cells via SLC6A14-Dependent Amino Acid Uptake. Implications for Treatment of Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 61, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Kontakioti, E.; Domvri, K.; Papakosta, D.; Daniilidis, M. HLA and asthma phenotypes/endotypes: A review. Hum. Immunol. 2014, 75, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.; Santiago, L.; Hutcheson, P.S.; Schwartz, H.J.; Spitznagel, E.; Castro, M.; Slavin, R.G.; Bellone, C.J. Evidence for the involvement of two different MHC class II regions in susceptibility or protection in allergic bronchopulmonary aspergillosis. J. Allergy Clin. Immunol. 2000, 106, 723–729. [Google Scholar] [CrossRef]
- Aron, Y.; Polla, B.S.; Bienvenu, T.; Dall’Ava, J.; Dusser, D.; Hubert, D. HLA Class II Polymorphism in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 1999, 159, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Hancock, D.B.; Artigas, M.S.; Gharib, S.A.; Henry, A.; Manichaikul, A.; Ramasamy, A.; Loth, D.W.; Imboden, M.; Koch, B.; McArdle, W.L.; et al. Genome-Wide Joint Meta-Analysis of SNP and SNP-by-Smoking Interaction Identifies Novel Loci for Pulmonary Function. PLoS Genet. 2012, 8, e1003098. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, W.K.; Gallins, P.; Pace, R.G.; Dang, H.; Wolf, W.E.; Jones, L.C.; Guo, X.; Zhou, Y.-H.; Madar, V.; Huang, J.; et al. Gene Expression in Transformed Lymphocytes Reveals Variation in Endomembrane and HLA Pathways Modifying Cystic Fibrosis Pulmonary Phenotypes. Am. J. Hum. Genet. 2015, 96, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, F.; Van Barneveld, A.; Hedtfeld, S.; Wölfl, S.; Becker, T.; Tümmler, B. The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur. J. Hum. Genet. 2013, 22, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Lee, H.J.; Kim, H.J.; Hong, S.H.; Pyo, J.O.; Cho, C.; Jung, Y.-K. Suppression of hypoxic cell death by APIP-induced sustained activation of AKT and ERK1. Oncogene 2007, 26, 2809–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, W.; Hong, S.H.; Lee, H.M.; Kim, N.Y.; Lim, Y.C.; Le, L.T.M.; Lim, B.; Kim, H.C.; Kim, T.Y.; Ashida, H.; et al. Structural and biochemical basis for the inhibition of cell death by APIP, a methionine salvage enzyme. Proc. Natl. Acad. Sci. USA 2013, 111, E54–E61. [Google Scholar] [CrossRef] [Green Version]
- Fossum, S.L.; Mutolo, M.J.; Tugores, A.; Ghosh, S.; Randell, S.H.; Jones, L.C.; Leir, S.-H.; Harris, A. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem. 2017, 292, 10938–10949. [Google Scholar] [CrossRef] [Green Version]
- Emond, M.J.; Louie, T.; Emerson, J.; Zhao, W.; Mathias, R.A.; Knowles, M.R.; Wright, F.A.; Rieder, M.J.; Tabor, H.K.; Nickerson, D.A.; et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet. 2012, 44, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Emond, M.J.; Louie, T.; Emerson, J.; Chong, J.X.; Mathias, R.A.; Knowles, M.R.; Rider, M.J.; Tabor, H.K.; Nickerson, D.A.; Barnes, K.C.; et al. Exome Sequencing of Phenotypic Extremes Identifies CAV2 and TMC6 as Interacting Modifiers of Chronic Pseudomonas aeruginosa Infection in Cystic Fibrosis. PLoS Genet 2015, 11, e1005273. [Google Scholar]
- Viel, M.; Hubert, D.; Burgel, P.R.; Génin, E.; Honoré, I.; Martinez, B.; Gaitch, N.; Chapron, J.; Kanaan, R.; Dusser, D.; et al. DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Clin. Respir. J. 2016, 10, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Schechter, M.S.; Shelton, B.J.; Margolis, P.A.; Fitzsimmons, S.C. The Association of Socioeconomic Status with Outcomes in Cystic Fibrosis Patients in the United States. Am. J. Respir. Crit. Care Med. 2001, 163, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Robinson, D.C.; Smyth, R.; Diggle, P.J.; Whitehead, M. A Longitudinal Study of the Impact of Social Deprivation and Disease Severity on Employment Status in the UK Cystic Fibrosis Population. PLoS ONE 2013, 8, e73322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Robinson, D.C.; Thielen, K.; Pressler, T.; Olesen, H.V.; Diderichsen, F.; Diggle, P.J.; Smyth, R.L.; Whitehead, M. Low socioeconomic status is associated with worse lung function in the Danish cystic fibrosis population. Eur. Respir. J. 2014, 44, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.T.; Ortega-García, J.A.; Sadreameli, S.C.; Wellmerling, J.; Cormet-Boyaka, E.; Thompson, R.; McGrath-Morrow, S.; Groner, J.A. The Impact of Secondhand Smoke Exposure on Children with Cystic Fibrosis: A Review. Int. J. Environ. Res. Public Heal. 2016, 13, 1003. [Google Scholar] [CrossRef]
- Brugha, R.; Edmondson, C.; Davies, J.C. Outdoor air pollution and cystic fibrosis. Paediatr. Respir. Rev. 2018, 28, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Goeminne, P.C.; Nawrot, T.S.; De Boeck, K.; Nemery, B.; Dupont, L.J. Proximity to blue spaces and risk of infection with Pseudomonas aeruginosa in cystic fibrosis: A case–control analysis. J. Cyst. Fibros. 2015, 14, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Daniels, T.; Goodacre, L.; Sutton, C.; Pollard, K.; Conway, S.; Peckham, D. Accurate assessment of adherence: Self-report and clinician report vs electronic monitoring of nebulizers. Chest 2011, 140, 425–432. [Google Scholar] [CrossRef]
- O’Donohoe, R.; Fullen, B.M. Adherence of Subjects with Cystic Fibrosis to Their Home Program: A Systematic Review. Respir. Care 2014, 59, 1731–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.; Mainz, J.G.; Gala, S.; Tabori, H.; Grossoehme, D. Adherence to therapies in cystic fibrosis: A targeted literature review. Expert Rev. Respir. Med. 2017, 11, 129–145. [Google Scholar] [CrossRef]
- Briesacher, B.A.; Quittner, A.L.; Saiman, L.; Sacco, P.; Fouayzi, H.; Quittell, L.M. Adherence with tobramycin inhaled solution and health care utilization. BMC Pulm. Med. 2011, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Nasr, S.Z.; Chou, W.; Villa, K.F.; Chang, E.; Broder, M.S. Adherence to dornase alfa treatment among commercially insured patients with cystic fibrosis. J. Med. Econ. 2013, 16, 801–808. [Google Scholar] [CrossRef]
- Quittner, A.L.; Zhang, J.; Marynchenko, M.; Chopra, P.A.; Signorovitch, J.; Yushkina, Y.; Riekert, K.A. Pulmonary Medication Adherence and Health-care Use in Cystic Fibrosis. Chest 2014, 146, 142–151. [Google Scholar] [CrossRef]
- Floch, J.; Ji, X.; Eakin, M.; Rudolf, I.; Pieper, K.; Nolte, H.; Junge, S.; Dopfer, C.; Sauer-Heilborn, A.; Ringshausen, F.C.; et al. Assessment of a Mobile App by Adolescents and Young Adults with Cystic Fibrosis: Pilot Evaluation. Jmir Mhealth Uhealth 2019, 7, e12442. [Google Scholar] [CrossRef] [Green Version]
- Bishay, L.C.; Sawicki, G.S. Strategies to optimize treatment adherence in adolescent patients with cystic fibrosis. Adolesc. Heal. Med. 2016, 7, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Einarsson, G.G.; Ronan, N.J.; Mooney, D.; McGettigan, C.; Mullane, D.; NiChroinin, M.; Shanahan, F.; Murphy, D.M.; McCarthy, M.; McCarthy, Y.; et al. Extended-culture and culture-independent molecular analysis of the airway microbiota in cystic fibrosis following CFTR modulation with ivacaftor. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef]
- Malsagova, K.A.; Butkova, T.V.; Kopylov, A.T.; Izotov, A.A.; Potoldykova, N.V.; Enikeev, D.V.; Grigoryan, V.; Tarasov, A.; Stepanov, A.A.; Kaysheva, A.L. Pharmacogenetic Testing: A Tool for Personalized Drug Therapy Optimization. Pharmaceutics 2020, 12, 1240. [Google Scholar] [CrossRef]
- Corvol, H.; Mésinèle, J.; Douksieh, I.-H.; Strug, L.J.; Boëlle, P.-Y.; Guillot, L. SLC26A9 Gene Is Associated with Lung Function Response to Ivacaftor in Patients with Cystic Fibrosis. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Kmit, A.; Marson, F.A.L.; Pereira, S.V.; Vinagre, A.M.; Leite, G.S.; Servidoni, M.F.; Ribeiro, J.D.; Ribeiro, A.F.; Bertuzzo, C.S.; Amaral, M.D. Extent of Rescue of F508del-CFTR Function by VX-809 and VX-770 in Human Nasal Epithelial Cells Correlates with SNP rs7512462 in SLC26A9 Gene in F508del/F508del Cystic Fibrosis Patients. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1323–1331. [Google Scholar] [CrossRef]
- Baker, E.; Harris, W.T.; Rowe, S.M.; Rutland, S.B.; Oates, G.R. Tobacco smoke exposure limits the therapeutic benefit of tezacaftor/ivacaftor in pediatric patients with cystic fibrosis. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, C.M.; Ryan, J.L.; Burns, L.; Wang, Y.; Zhang, N.; Clancy, J.P.; Drotar, D. Electronic monitoring reveals highly variable adherence patterns in patients prescribed ivacaftor. J. Cyst. Fibros. 2015, 14, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Stanton, B.A.; Coutermarsh, B.; Barnaby, R.; Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PLoS ONE 2015, 10, e0127742. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Aksit, M.A.; Pace, R.G.; Vecchio-Pagán, B.; Ling, H.; Rommens, J.M.; Boelle, P.-Y.; Guillot, L.; Raraigh, K.S.; Pugh, E.; Zhang, P.; et al. Genetic Modifiers of Cystic Fibrosis-Related Diabetes Have Extensive Overlap with Type 2 Diabetes and Related Traits. J. Clin. Endocrinol. Metab. 2019, 105, 1401–1415. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Class of Mutation | Impact on CFTR Protein and Example Mutations |
|---|---|
| I | Defect in protein synthesis, e.g., G542X, R553X, R1162X, W1282X |
| II | Defect in protein trafficking to cell membrane, e.g., G85E, I507del, F508del, N1303K |
| III | CFTR protein reaches the cell membrane but defect in channel gating, e.g., S549R, G551D, G1349D |
| IV | CFTR protein reaches the cell membrane but there is defective conductance, e.g., R117H, R334W, D1152H |
| V | Normal folding of the CFTR protein with normal function, but amount of protein made is insufficient, e.g., A455E, 2789+5G>A, 3849+10kbC>T |
| VI | CFTR protein is produced and is in the correct location but has reduced stability with rapid turnover, e.g., F508del, Q1411X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes 2021, 12, 562. https://doi.org/10.3390/genes12040562
Sepahzad A, Morris-Rosendahl DJ, Davies JC. Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes. 2021; 12(4):562. https://doi.org/10.3390/genes12040562
Chicago/Turabian StyleSepahzad, Afsoon, Deborah J. Morris-Rosendahl, and Jane C. Davies. 2021. "Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine" Genes 12, no. 4: 562. https://doi.org/10.3390/genes12040562
APA StyleSepahzad, A., Morris-Rosendahl, D. J., & Davies, J. C. (2021). Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes, 12(4), 562. https://doi.org/10.3390/genes12040562