
OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters

 , , ,
, , ,  , and
, and
Abstract
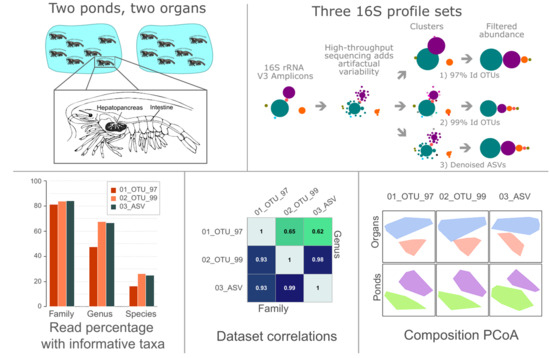
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, 16S rDNA Amplicon Preparation, and Sequencing
2.3. Data Preprocessing
2.4. OTU Clustering (Identity-Based)
2.5. ASV Clustering (Denoising)
2.6. Chimera Filtering, Taxonomic Identification, and Filters
2.7. Comparing the Performance of OTU and ASV Sets
2.8. α-Diversity Comparison (Within-Sample)
2.9. β-Diversity Comparison (Between-Sample)
3. Results
3.1. Different Preprocessing and Clustering Methods Produced Distinct Sets of Clusters
3.2. Sequence-Level Analyses Show Well-Outlined ASV Clusters and Partially Clusterable OTU Sets That Are Origin-Dependent
3.3. Filters to Retain OTUs and ASVs, Accounting for >0.1% of the Total Abundance Per Sample
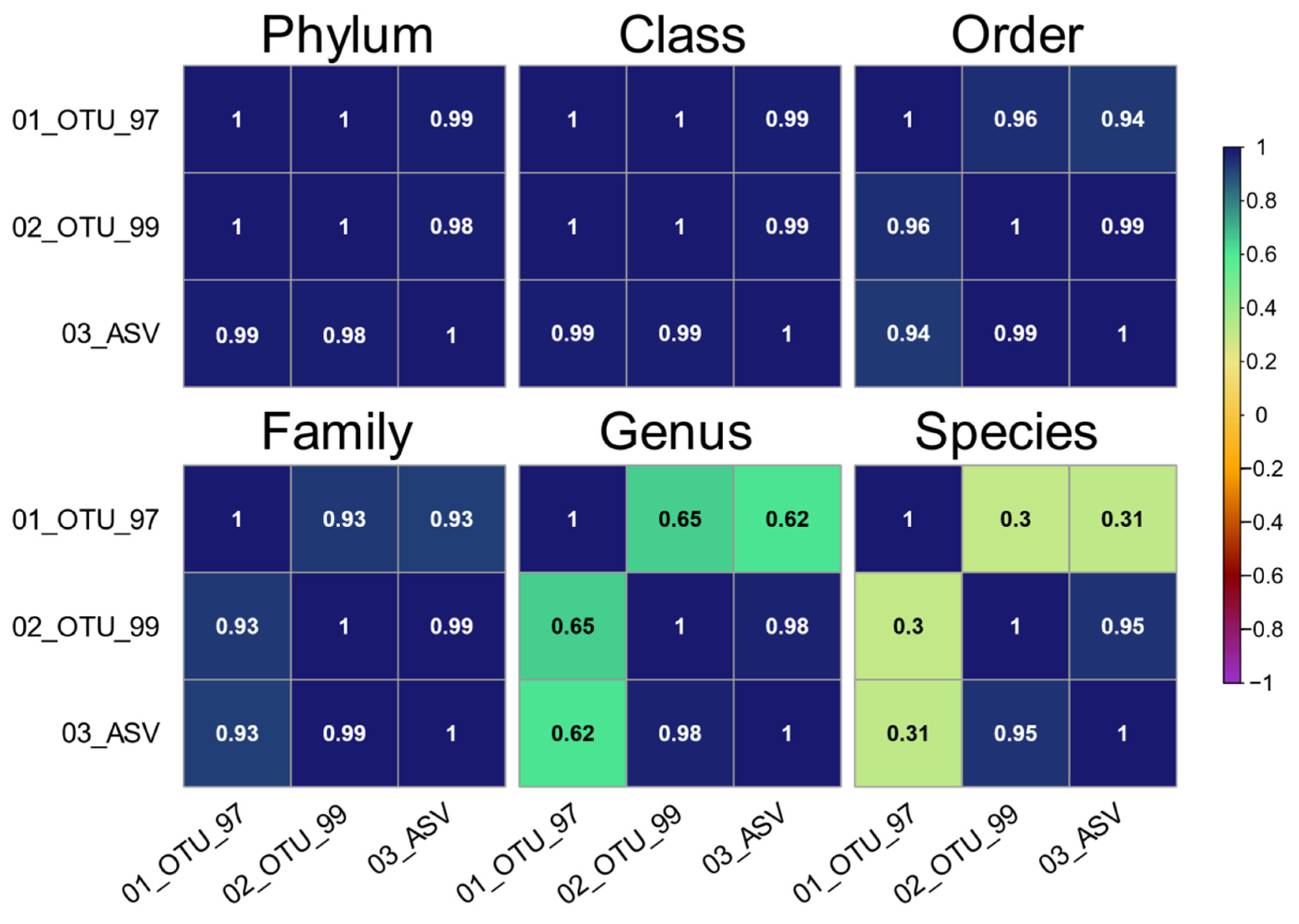
3.4. Evaluating Taxonomy-Related Differences
3.5. Collated Group Richness and Entropy Evaluated through α-Diversity
3.6. Group Abundance and Composition Differences Evaluated through β-Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2020, 1st ed.; Food and Agriculture Organization of the United Nations, Ed.; FAO: Rome, Italy, 2020; ISBN 978-92-5-132692-3. [Google Scholar]
- FAO. Introductions and Movement of Penaeus Vannamei and Penaeus Stylirostris in Asia and the Pacific; FAO: Bangkok, Thailand, 2004. [Google Scholar]
- Ghaffari, N.; Sanchez-Flores, A.; Doan, R.; Garcia-Orozco, K.D.; Chen, P.L.; Ochoa-Leyva, A.; Lopez-Zavala, A.A.; Carrasco, J.S.; Hong, C.; Brieba, L.G.; et al. Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci. Rep. 2014, 4, 7081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornejo-Granados, F.; Lopez-Zavala, A.A.; Gallardo-Becerra, L.; Mendoza-Vargas, A.; Sánchez, F.; Vichido, R.; Brieba, L.G.; Viana, M.T.; Sotelo-Mundo, R.R.; Ochoa-Leyva, A. Microbiome of Pacific Whiteleg shrimp reveals differential bacterial community composition between Wild, Aquacultured and AHPND/EMS outbreak conditions. Sci. Rep. 2017, 7, 11783. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wang, Y.; Liu, Q.; Xiong, D.; Zhang, J. Transcriptomic and microbiota response on Litopenaeus vannamei intestine subjected to acute sulfide exposure. Fish Shellfish Immunol. 2019, 88, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Perez-Enriquez, R.; Hernández-Martínez, F.; Cruz, P. Genetic diversity status of White shrimp Penaeus (Litopenaeus) vannamei broodstock in Mexico. Aquaculture 2009, 297, 44–50. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 2017, 19, 1490–1501. [Google Scholar] [CrossRef]
- Dai, W.F.; Zhang, J.J.; Qiu, Q.F.; Chen, J.; Yang, W.; Ni, S.; Xiong, J.B. Starvation stress affects the interplay among shrimp gut microbiota, digestion, and immune activities. Fish Shellfish Immunol. 2018, 80, 191–199. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, Y.; Chen, K.; Yu, N.; Zhou, Z.; Chen, L.; Du, Z.; Li, E. Characterization of the intestinal microbiota in Pacific white shrimp, Litopenaeus vannamei, fed diets with different lipid sources. Aquaculture 2014, 434, 449–455. [Google Scholar] [CrossRef]
- García-López, R.; Cornejo-Granados, F.; Lopez-Zavala, A.A.; Sánchez-López, F.; Cota-Huízar, A.; Sotelo-Mundo, R.R.; Guerrero, A.; Mendoza-Vargas, A.; Gómez-Gil, B.; Ochoa-Leyva, A. Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, R79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R.; et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Glassman, S.I.; Martiny, J.B.H. Broadscale Ecological Patterns Are Robust to Use of Exact. mSphere 2018, 3, e00148-18. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, S.; Atakora, F.; Fehr, K.; Khafipour, E. Biological observations in microbiota analysis are robust to the choice of 16S rRNA gene sequencing processing algorithm: Case study on human milk microbiota. BMC Microbiol. 2020, 20, 290. [Google Scholar] [CrossRef] [PubMed]
- Caruso, V.; Song, X.; Asquith, M.; Karstens, L. Performance of Microbiome Sequence Inference Methods in Environments with Varying Biomass. mSystems 2019, 4, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Md Zoqratt, M.Z.H.; Eng, W.W.H.; Thai, B.T.; Austin, C.M.; Gan, H.M. Microbiome analysis of Pacific white shrimp gut and rearing water from Malaysia and Vietnam: Implications for aquaculture research and management. PeerJ 2018, 6, e5826. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Farfante Perez, I.; Frederick Kensley, B. Penaeoid and Sergestoid Shrimps and Prawns of the World: Keys and Diagnoses for the Families and Genera, 1st ed.; Editions du Muséum: Paris, France, 1997; ISBN 2856535100. [Google Scholar]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Yuan, J.; Yiu, S.M.; Li, Z.; Xie, Y.; Chen, Y.; Shi, Y.; Zhang, H.; Li, Y.; Lam, T.W.; et al. COPE: An accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics 2012, 28, 2870–2874. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 23 May 2020).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Kyrpides, N.C. Genomes Online Database (GOLD 1.0): A monitor of complete and ongoing genome projects worldwide. Bioinformatics 1999, 15, 773–774. [Google Scholar] [CrossRef] [PubMed]
- Varoquaux, G.; Buitinck, L.; Louppe, G.; Grisel, O.; Pedregosa, F.; Mueller, A. Scikit-learn: Machine Learning without Learning the Machinery. GetMobile Mob. Comput. Commun. 2015, 19, 29–33. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Jari Oksanen, F.; Guillaume, B.; Michael, F.; Roeland, K.; Pierre, L.; Dan McGlinn, P.R.; Minchin, R.B.; O’Hara, G.L.; Simpson, P.; Solymos, M.; et al. The Vegan Community Ecology Package. 2019. Available online: https://cran.r-project.org/web/packages/vegan/ (accessed on 23 May 2020).
- Janssen, S.; Mcdonald, D.; Gonzalez, A.; Navas-molina, J.A.; Jiang, L.; Xu, Z. Phylogenetic Placement of Exact Amplicon Sequences. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M.; Hou, Z.; Yuan, J.; Liu, Y.; Qu, Y.; Liu, B. Taxonomic and functional metagenomic profiling of gastrointestinal tract microbiome of the farmed adult turbot (Scophthalmus maximus). FEMS Microbiol. Ecol. 2013, 86, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Schryver, P.; Vadstein, O. Ecological theory as a foundation to control pathogenic invasion in aquaculture. ISME J. 2014, 8, 2360–2368. [Google Scholar] [CrossRef] [Green Version]
- Cornejo-Granados, F.; Gallardo-Becerra, L.; Leonardo-Reza, M.; Ochoa-Romo, J.P.; Ochoa-Leyva, A. A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 2018, 6, e5382. [Google Scholar] [CrossRef]
- Cheung, M.K.; Yip, H.Y.; Nong, W.; Law, P.T.W.; Chu, K.H.; Kwan, H.S.; Hui, J.H.L. Rapid Change of Microbiota Diversity in the Gut but Not the Hepatopancreas During Gonadal Development of the New Shrimp Model Neocaridina denticulata. Mar. Biotechnol. 2015, 17, 811–819. [Google Scholar] [CrossRef]
- Chen, T.; Wong, N.K.; Jiang, X.; Luo, X.; Zhang, L.; Yang, D.; Ren, C.; Hu, C. Nitric oxide as an antimicrobial molecule against Vibrio harveyi infection in the hepatopancreas of Pacific white shrimp, Litopenaeus vannamei. Fish Shellfish Immunol. 2015, 42, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Wu, X.; Wu, W.; Dai, C.; Yuan, J. Ferritin protect shrimp Litopenaeus vannamei from WSSV infection by inhibiting virus replication. Fish Shellfish Immunol. 2015, 42, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Chen, L.; Ding, Z.; Qin, J.; Sun, S.; Wang, L.; Ye, J. Molecular Cloning, Characterization, and mRNA Expression of Hemocyanin Subunit in Oriental River Prawn Macrobrachium nipponense. Int. J. Genom. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.D.; Fu, L.D.; Jia, Y.P.; Du, X.J.; Wang, Q.; Wang, Y.H.; Zhao, X.F.; Yu, X.Q.; Wang, J.X. A hepatopancreas-specific C-type lectin from the Chinese shrimp Fenneropenaeus chinensis exhibits antimicrobial activity. Mol. Immunol. 2008, 45, 348–361. [Google Scholar] [CrossRef]
- Sze, M.A.; Schloss, P.D. The Impact of DNA Polymerase and Number of Rounds of Amplification in PCR on 16S rRNA Gene Sequence Data. mSphere 2019, 4, e00163-19. [Google Scholar] [CrossRef] [Green Version]
- May, A.; Abeln, S.; Buijs, M.J.; Heringa, J.; Crielaard, W.; Brandt, B.W. NGS-eval: NGS error analysis and novel sequence VAriant detection tooL. Nucleic Acids Res. 2015, 43, W301–W305. [Google Scholar] [CrossRef]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Balebona, M.C.; Andreu, M.J.; Bordas, M.A.; Zorilla, I.; Moriñgo, M.A.; Borrego, J.J. Pathogenicity of Vibrio alginolyticus for cultured gilt-head sea bream (Sparus aurata L.). Appl. Environ. Microbiol. 1998, 64, 4269–4275. [Google Scholar] [CrossRef] [Green Version]
- Borrego, J.J.; Castro, D.; Luque, A.; Paillard, C.; Maes, P.; Garcia, M.T.; Ventosa, A. Vibrio tapetis sp. nov., the causative agent of the brown ring disease affecting cultured clams. Int. J. Syst. Bacteriol. 1996, 46, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Karoonuthaisiri, N. Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. J. Invertebr. Pathol. 2016, 133, 12–19. [Google Scholar] [CrossRef]
- Tran, L.; Nunan, L.; Redman, R.M.; Mohney, L.L.; Pantoja, C.R.; Fitzsimmons, K.; Lightner, D.V. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis. Aquat. Organ. 2013, 105, 45–55. [Google Scholar] [CrossRef]
- De la pena, L.D.; Nakai, T.; Muroga, K.; Momoyama, K. Detection of the Causative Bacterium of Vibriosis in Kuruma Prawn, Penaeus japonicus. Fish Pathol. 1992, 27, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Chen, P.C.; Weng, F.C.H.; Shaw, G.T.W.; Wang, D. Habitat and indigenous gut microbes contribute to the plasticity of gut microbiome in oriental river prawn during rapid environmental change. PLoS ONE 2017, 12, e0181427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Li, W.J.; Zhang, K.Y.; Tian, X.P.; Jiang, Y.; Xu, L.H.; Jiang, C.L.; Lai, R. Massilia dura sp. nov., Massilia albidiflava sp. nov., Massilia plicata sp. nov. and Massilia lutea sp. nov., isolated from soils in China. Int. J. Syst. Evol. Microbiol. 2006, 56, 459–463. [Google Scholar] [CrossRef]
- Faramarzi, M.; Fazeli, M.; Tabatabaei, M.; Adrangi, S.; Jami Al Ah, K.; Tasharrofi, N.; Aziz Mohse, F. Optimization of Cultural Conditions for Production of Chitinase by a Soil Isolate of Massilia timonae. Biotechnology 2009, 8, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Wei, D.; Deng, X.; Weng, S.; He, Z.; He, J. Environmental factors shape water microbial community structure and function in shrimp cultural enclosure ecosystems. Front. Microbiol. 2017, 8, 2359. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Hameed, A.; Arun, A.B.; Liu, Y.C.; Hsu, Y.H.; Lai, W.A.; Rekha, P.D.; Young, C.C. Description of Noviherbaspirillum malthae gen. nov., sp. nov., isolated from an oil-contaminated soil, and proposal to reclassify herbaspirillum soli, Herbaspirillum aurantiacum, Herbaspirillum canariense and Herbaspirillum psychrotolerans as Noviherbaspi. Int. J. Syst. Evol. Microbiol. 2013, 63, 4100–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.B.; Nie, L.; Chen, J. Current understanding on the roles of gut microbiota in fish disease and immunity. Zool. Res. 2019, 40, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Hou, D.; Zhou, R.; Zeng, S.; Xing, C.; Wei, D.; Deng, X.; Yu, L.; Wang, H.; Deng, Z.; et al. Sediment microbial communities contribute to shrimp intestine microbiota in cultural pond ecosystems. Gen. Microbiol. 2020, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Zhu, J.; Zhang, D. The application of bacterial indicator phylotypes to predict shrimp health status. Appl. Microbiol. Biotechnol. 2014, 98, 8291–8299. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, X.; Xiong, J.; Zhu, J.; Wang, Y.; Zhao, Q.; Chen, H.; Guo, A.; Wu, J.; Dai, H. Bacterioplankton assemblages as biological indicators of shrimp health status. Ecol. Indic. 2014, 38, 218–224. [Google Scholar] [CrossRef]
- Gonçalves, A.T.; Collipal-Matamal, R.; Valenzuela-Muñoz, V.; Nuñez-Acuña, G.; Valenzuela-Miranda, D.; Gallardo-Escárate, C. Nanopore sequencing of microbial communities reveals the potential role of sea lice as a reservoir for fish pathogens. Sci. Rep. 2020, 10, 2895. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Set | Org R | Pond R | Org-Pond R | Org Pval | Pond Pval | Org-Pond Pval |
|---|---|---|---|---|---|---|---|
| Unweighted Unifrac | 01_OTU_97 | 0.297 | 0.336 | 0.802 | 0.006 | 0.001 | 0.001 |
| Unweighted Unifrac | 02_OTU_99 | 0.273 | 0.352 | 0.805 | 0.01 | 0.001 | 0.001 |
| Unweighted Unifrac | 03_ASV | 0.226 | 0.294 | 0.692 | 0.013 | 0.002 | 0.001 |
| Weighted Unifrac | 01_OTU_97 | 0.168 | 0.165 | 0.607 | 0.022 | 0.031 | 0.001 |
| Weighted Unifrac | 02_OTU_99 | 0.166 | 0.153 | 0.592 | 0.028 | 0.034 | 0.001 |
| Weighted Unifrac | 03_ASV | 0.159 | 0.159 | 0.596 | 0.022 | 0.028 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-López, R.; Cornejo-Granados, F.; Lopez-Zavala, A.A.; Cota-Huízar, A.; Sotelo-Mundo, R.R.; Gómez-Gil, B.; Ochoa-Leyva, A. OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters. Genes 2021, 12, 564. https://doi.org/10.3390/genes12040564
García-López R, Cornejo-Granados F, Lopez-Zavala AA, Cota-Huízar A, Sotelo-Mundo RR, Gómez-Gil B, Ochoa-Leyva A. OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters. Genes. 2021; 12(4):564. https://doi.org/10.3390/genes12040564
Chicago/Turabian StyleGarcía-López, Rodrigo, Fernanda Cornejo-Granados, Alonso A. Lopez-Zavala, Andrés Cota-Huízar, Rogerio R. Sotelo-Mundo, Bruno Gómez-Gil, and Adrian Ochoa-Leyva. 2021. "OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters" Genes 12, no. 4: 564. https://doi.org/10.3390/genes12040564
APA StyleGarcía-López, R., Cornejo-Granados, F., Lopez-Zavala, A. A., Cota-Huízar, A., Sotelo-Mundo, R. R., Gómez-Gil, B., & Ochoa-Leyva, A. (2021). OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters. Genes, 12(4), 564. https://doi.org/10.3390/genes12040564