
Application of Genome Sequencing from Blood to Diagnose Mitochondrial Diseases

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Genome Sequencing Detects Low Levels of mtDNA Heteroplasmy in Blood
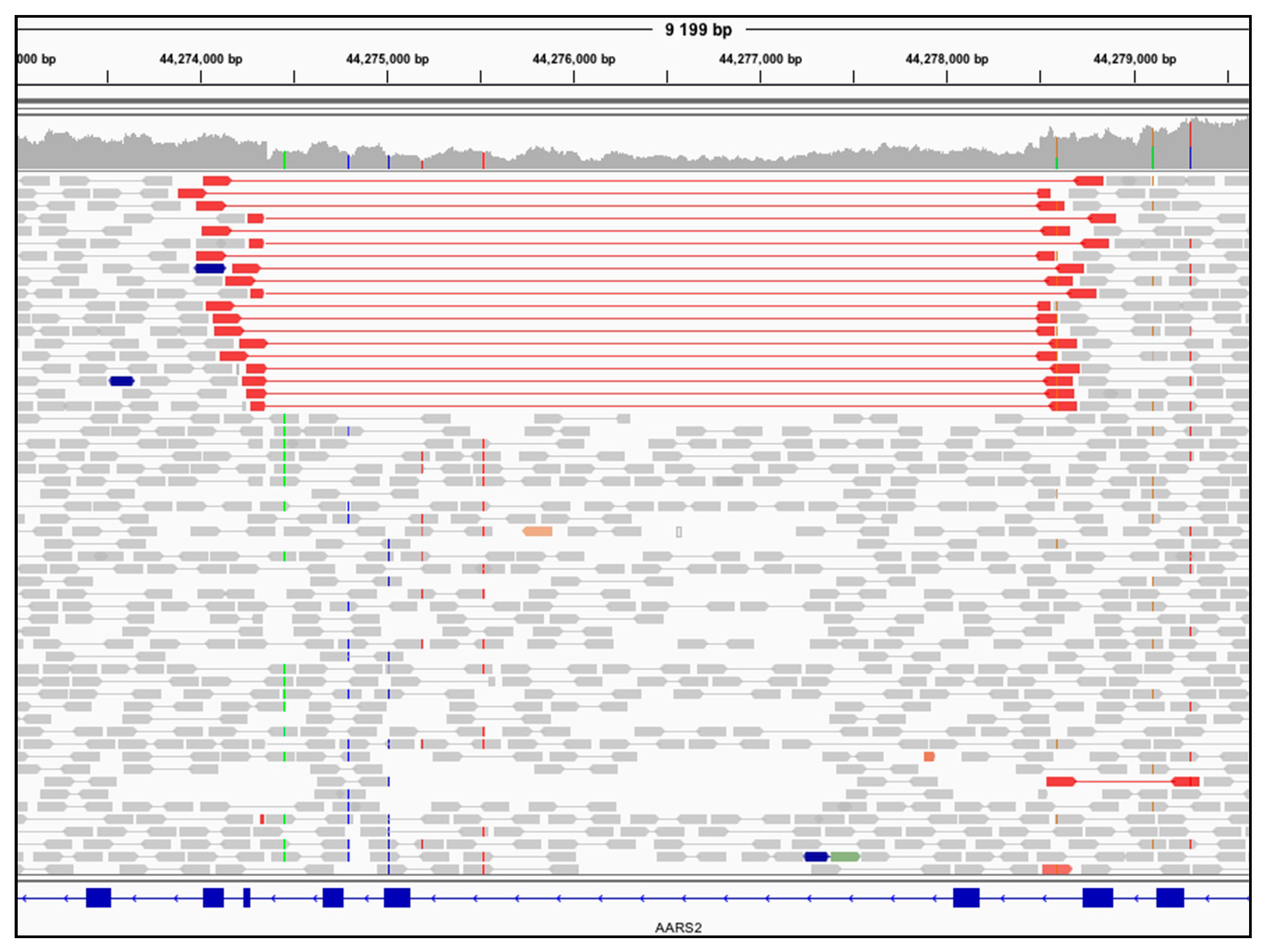
3.2. Genome Sequencing Detects an Intragenic Nuclear DNA Deletion
3.3. Genome Sequencing Detects a Large Mitochondrial DNA Deletion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Haberberger, B.; Frisch, E.M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.W.; Pyle, A.; Griffin, H.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.L.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, A.; Murayama, K.; Mori, M.; Harashima, H.; Yamazaki, T.; Tamaru, S.; Yamashita, Y.; Kishita, Y.; Nakachi, Y.; Kohda, M.; et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: Exome sequencing for disease gene identification. Biochim. Biophys. Acta 2014, 1840, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta. 2016, 1857, 1326–1335. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Puusepp, S.; Reinson, K.; Pajusalu, S.; Murumets, U.; Oiglane-Shlik, E.; Rein, R.; Talvik, I.; Rodenburg, R.J.; Ounap, K. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol. Genet. Metab. Rep. 2018, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.E.J.; Nguyen, M.; Kamps, R.; Hendrickx, A.T.; Sallevelt, S.; Gottschalk, R.W.H.; Calis, C.M.; Stassen, A.P.M.; de Koning, B.; Mulder-Den Hartog, E.N.M.; et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front. Genet. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef]
- Stark, Z.; Boughtwood, T.; Phillips, P.; Christodoulou, J.; Hansen, D.P.; Braithwaite, J.; Newson, A.J.; Gaff, C.L.; Sinclair, A.H.; North, K.N. Australian genomics: A federated model for integrating genomics into healthcare. Am. J. Hum. Genet. 2019, 105, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef] [PubMed]
- Gayevskiy, V.; Roscioli, T.; Dinger, M.E.; Cowley, M.J. Seave: A comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics 2019, 35, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Puttick, C.; Kumar, K.R.; Davis, R.L.; Pinese, M.; Thomas, D.M.; Dinger, M.E.; Sue, C.M.; Cowley, M.J. Mity: A highly sensitive mitochondrial variant analysis pipeline for whole genome sequencing data. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Minoche, A.E.; Lundie, B.; Peters, G.B.; Ohnesorg, T.; Pinese, M.; Thomas, D.M.; Zankl, A.; Roscioli, T.; Schonrock, N.; Kummerfeld, S.; et al. ClinSV: Clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med. 2021, 13, 32. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. PON-mt-tRNA: A multifactorial probability-based method for classification of mitochondrial tRNA variations. Nucleic Acids Res. 2016, 44, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. HmtVar: A new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Akesson, L.S.; Eggers, S.; Love, C.J.; Chong, B.; Krzesinski, E.I.; Brown, N.J.; Tan, T.Y.; Richmond, C.M.; Thorburn, D.R.; Christodoulou, J.; et al. Early diagnosis of Pearson syndrome in neonatal intensive care following rapid mitochondrial genome sequencing in tandem with exome sequencing. Eur. J. Hum. Genet. 2019, 27, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Kukat, A.; Kukat, C.; Brocher, J.; Schafer, I.; Krohne, G.; Trounce, I.A.; Villani, G.; Seibel, P. Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res. 2008, 36, e44. [Google Scholar] [CrossRef] [Green Version]
- Lake, N.J.; Webb, B.D.; Stroud, D.A.; Richman, T.R.; Ruzzenente, B.; Compton, A.G.; Mountford, H.S.; Pulman, J.; Zangarelli, C.; Rio, M.; et al. Biallelic Mutations in MRPS34 lead to instability of the small mitoribosomal subunit and leigh syndrome. Am. J. Hum. Genet. 2017, 101, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohlin, A.; Wernersson, J.; Engwall, Y.; Wiklund, L.; Bjork, J.; Nordling, M. Parallel sequencing used in detection of mosaic mutations: Comparison with four diagnostic DNA screening techniques. Hum. Mutat. 2009, 30, 1012–1020. [Google Scholar] [CrossRef]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putz, J.; Dupuis, B.; Sissler, M.; Florentz, C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 2007, 13, 1184–1190. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Alston, C.L.; Bender, A.; Hargreaves, I.P.; Mundy, H.; Deshpande, C.; Klopstock, T.; McFarland, R.; Horvath, R.; Taylor, R.W. The pathogenic m.3243A>T mitochondrial DNA mutation is associated with a variable neurological phenotype. Neuromuscul. Disord. 2010, 20, 403–406. [Google Scholar] [CrossRef]
- Czell, D.; Abicht, A.; Hench, J.; Weber, M. Exercise-induced myalgia and rhabdomyolysis in a patient with the rare m.3243A>T mtDNA mutation. BMJ Case Rep. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Osaka, H.; Shimbo, H.; Tajika, M.; Yamazaki, M.; Ueda, A.; Murayama, K.; Yamagata, T. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum. Genome Var. 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Sohm, B.; Frugier, M.; Brule, H.; Olszak, K.; Przykorska, A.; Florentz, C. Towards understanding human mitochondrial leucine aminoacylation identity. J. Mol. Biol. 2003, 328, 995–1010. [Google Scholar] [CrossRef]
- Grady, J.P.; Pickett, S.J.; Ng, Y.S.; Alston, C.L.; Blakely, E.L.; Hardy, S.A.; Feeney, C.L.; Bright, A.A.; Schaefer, A.M.; Gorman, G.S.; et al. MtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Sommerville, E.W.; Zhou, X.L.; Olahova, M.; Jenkins, J.; Euro, L.; Konovalova, S.; Hilander, T.; Pyle, A.; He, L.; Habeebu, S.; et al. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Hum. Mol. Genet. 2019, 28, 258–268. [Google Scholar] [CrossRef]
- Srivastava, S.; Butala, A.; Mahida, S.; Richter, J.; Mu, W.; Poretti, A.; Vernon, H.; VanGerpen, J.; Atwal, P.S.; Middlebrooks, E.H.; et al. Expansion of the clinical spectrum associated with AARS2-related disorders. Am. J. Med. Genet. A 2019, 179, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Euro, L.; Konovalova, S.; Asin-Cayuela, J.; Tulinius, M.; Griffin, H.; Horvath, R.; Taylor, R.W.; Chinnery, P.F.; Schara, U.; Thorburn, D.R.; et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Hock, D.H.; Reljic, B.; Ang, C.S.; Muellner-Wong, L.; Mountford, H.S.; Compton, A.G.; Ryan, M.T.; Thorburn, D.R.; Stroud, D.A. HIGD2A is Required for Assembly of the COX3 Module of Human Mitochondrial Complex IV. Mol. Cell Proteom. 2020, 19, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014, 82, 2063–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Serrano, L.E.; Chihade, J.W.; Sissler, M. When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019, 294, 5309–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damas, J.; Carneiro, J.; Amorim, A.; Pereira, F. MitoBreak: The mitochondrial DNA breakpoints database. Nucleic Acids Res. 2014, 42, D1261–D1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broomfield, A.; Sweeney, M.G.; Woodward, C.E.; Fratter, C.; Morris, A.M.; Leonard, J.V.; Abulhoul, L.; Grunewald, S.; Clayton, P.T.; Hanna, M.G.; et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015, 38, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Sadikovic, B.; Wang, J.; El-Hattab, A.W.; Landsverk, M.; Douglas, G.; Brundage, E.K.; Craigen, W.J.; Schmitt, E.S.; Wong, L.J. Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes. PLoS ONE 2010, 5, e15687. [Google Scholar] [CrossRef]
- Lee, H.F.; Lee, H.J.; Chi, C.S.; Tsai, C.R.; Chang, T.K.; Wang, C.J. The neurological evolution of Pearson syndrome: Case report and literature review. Eur. J. Paediatr. Neurol. 2007, 11, 208–214. [Google Scholar] [CrossRef]
- Farruggia, P.; Di Cataldo, A.; Pinto, R.M.; Palmisani, E.; Macaluso, A.; Valvo, L.L.; Cantarini, M.E.; Tornesello, A.; Corti, P.; Fioredda, F.; et al. Pearson syndrome: A retrospective cohort study from the marrow failure study group of A.I.E.O.P. (Associazione Italiana Emato-Oncologia Pediatrica). JIMD Rep. 2016, 26, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, S.B.; Mayr, J.A.; Nuoffer, J.M.; Prokisch, H.; Sperl, W. A guideline for the diagnosis of pediatric mitochondrial disease: The value of muscle and skin biopsies in the genetics era. Neuropediatrics 2017, 48, 309–314. [Google Scholar] [CrossRef]
- Zambelli, F.; Vancampenhout, K.; Daneels, D.; Brown, D.; Mertens, J.; Van Dooren, S.; Caljon, B.; Gianaroli, L.; Sermon, K.; Voet, T.; et al. Accurate and comprehensive analysis of single nucleotide variants and large deletions of the human mitochondrial genome in DNA and single cells. Eur. J. Hum. Genet. 2017, 25, 1229–1236. [Google Scholar] [CrossRef]
- Grady, J.P.; Murphy, J.L.; Blakely, E.L.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Tuppen, H.A. Accurate measurement of mitochondrial DNA deletion level and copy number differences in human skeletal muscle. PLoS ONE 2014, 9, e114462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Li, F.; Chen, D.; Wang, G.; Truong, C.K.; Enns, G.M.; Graham, B.; Milone, M.; Landsverk, M.L.; Wang, J.; et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. 2013, 15, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadalayil, L.; Rafiq, S.; Rose-Zerilli, M.J.; Pengelly, R.J.; Parker, H.; Oscier, D.; Strefford, J.C.; Tapper, W.J.; Gibson, J.; Ennis, S.; et al. Exome sequence read depth methods for identifying copy number changes. Brief. Bioinform. 2015, 16, 380–392. [Google Scholar] [CrossRef]
- Gross, A.M.; Ajay, S.S.; Rajan, V.; Brown, C.; Bluske, K.; Burns, N.J.; Chawla, A.; Coffey, A.J.; Malhotra, A.; Scocchia, A.; et al. Copy-number variants in clinical genome sequencing: Deployment and interpretation for rare and undiagnosed disease. Genet. Med. 2019, 21, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghvi, R.V.; Buhay, C.J.; Powell, B.C.; Tsai, E.A.; Dorschner, M.O.; Hong, C.S.; Lebo, M.S.; Sasson, A.; Hanna, D.S.; McGee, S.; et al. Characterizing reduced coverage regions through comparison of exome and genome sequencing data across 10 centers. Genet. Med. 2018, 20, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Kohler, S.; Zemojtel, T.; Spielmann, M.; Jager, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, R.; Riley, L.G.; Guo, Y.; Menezes, M.; Compton, A.G.; Van Bergen, N.J.; Gayevskiy, V.; Cowley, M.J.; Cummings, B.B.; Adams, L.; et al. Cryptic intronic NBAS variant reveals the genetic basis of recurrent liver failure in a child. Mol. Genet. Metab. 2019, 126, 77–82. [Google Scholar] [CrossRef]
- Helman, G.; Compton, A.G.; Hock, D.H.; Walkiewicz, M.; Brett, G.R.; Pais, L.; Tan, T.Y.; De Paoli-Iseppi, R.; Clark, M.B.; Christodoulou, J.; et al. Multiomic analysis elucidates Complex I deficiency caused by a deep intronic variant in NDUFB10. Hum. Mutat. 2020, 42, 19–24. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rius, R.; Compton, A.G.; Baker, N.L.; Welch, A.E.; Coman, D.; Kava, M.P.; Minoche, A.E.; Cowley, M.J.; Thorburn, D.R.; Christodoulou, J. Application of Genome Sequencing from Blood to Diagnose Mitochondrial Diseases. Genes 2021, 12, 607. https://doi.org/10.3390/genes12040607
Rius R, Compton AG, Baker NL, Welch AE, Coman D, Kava MP, Minoche AE, Cowley MJ, Thorburn DR, Christodoulou J. Application of Genome Sequencing from Blood to Diagnose Mitochondrial Diseases. Genes. 2021; 12(4):607. https://doi.org/10.3390/genes12040607
Chicago/Turabian StyleRius, Rocio, Alison G. Compton, Naomi L. Baker, AnneMarie E. Welch, David Coman, Maina P. Kava, Andre E. Minoche, Mark J. Cowley, David R. Thorburn, and John Christodoulou. 2021. "Application of Genome Sequencing from Blood to Diagnose Mitochondrial Diseases" Genes 12, no. 4: 607. https://doi.org/10.3390/genes12040607
APA StyleRius, R., Compton, A. G., Baker, N. L., Welch, A. E., Coman, D., Kava, M. P., Minoche, A. E., Cowley, M. J., Thorburn, D. R., & Christodoulou, J. (2021). Application of Genome Sequencing from Blood to Diagnose Mitochondrial Diseases. Genes, 12(4), 607. https://doi.org/10.3390/genes12040607