
Molecular Insights into Mitochondrial Protein Translocation and Human Disease


Abstract
:1. Introduction
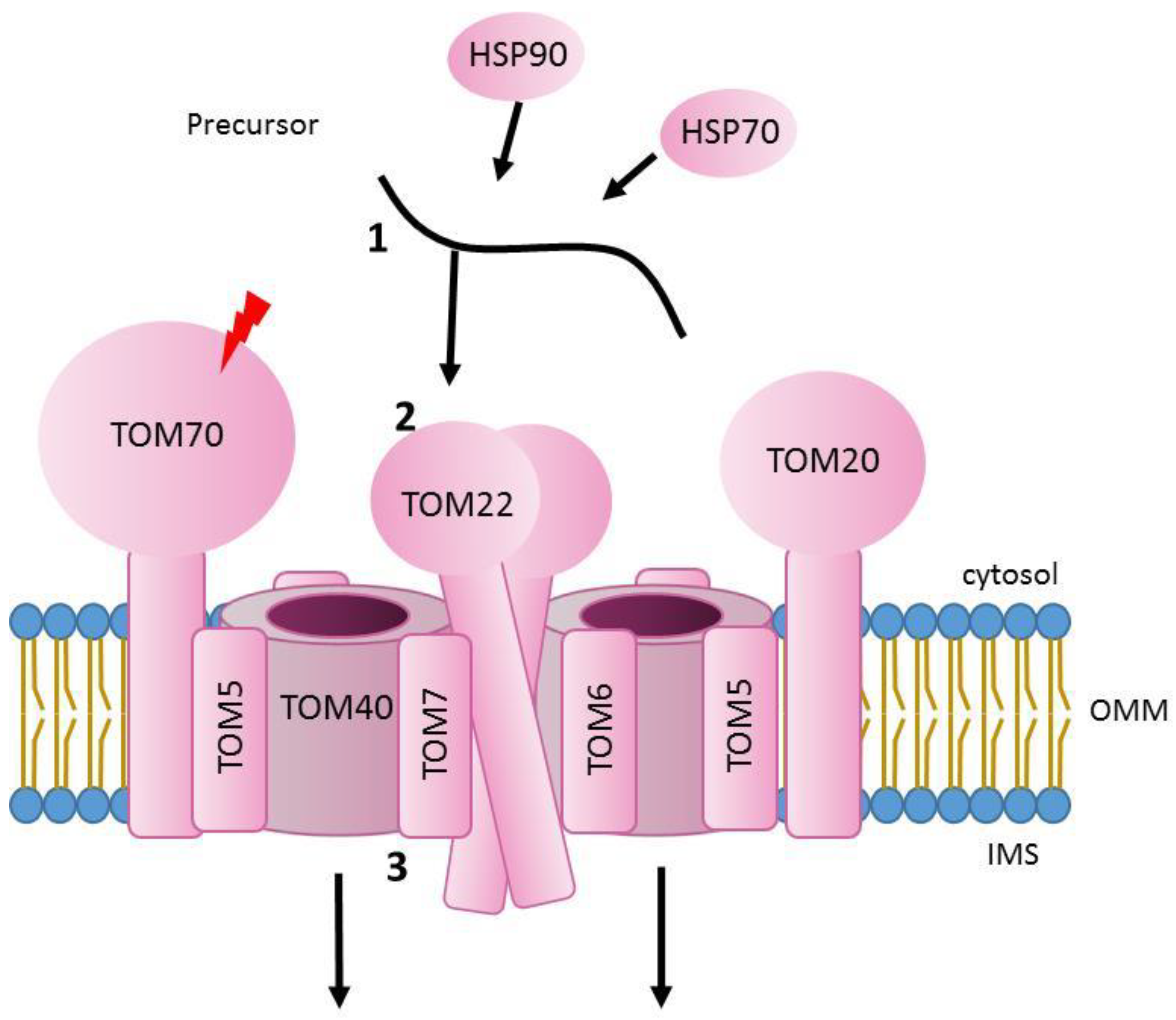
2. Translocation through the Outer Mitochondrial Membrane
3. Mutations Affecting TOM Complex
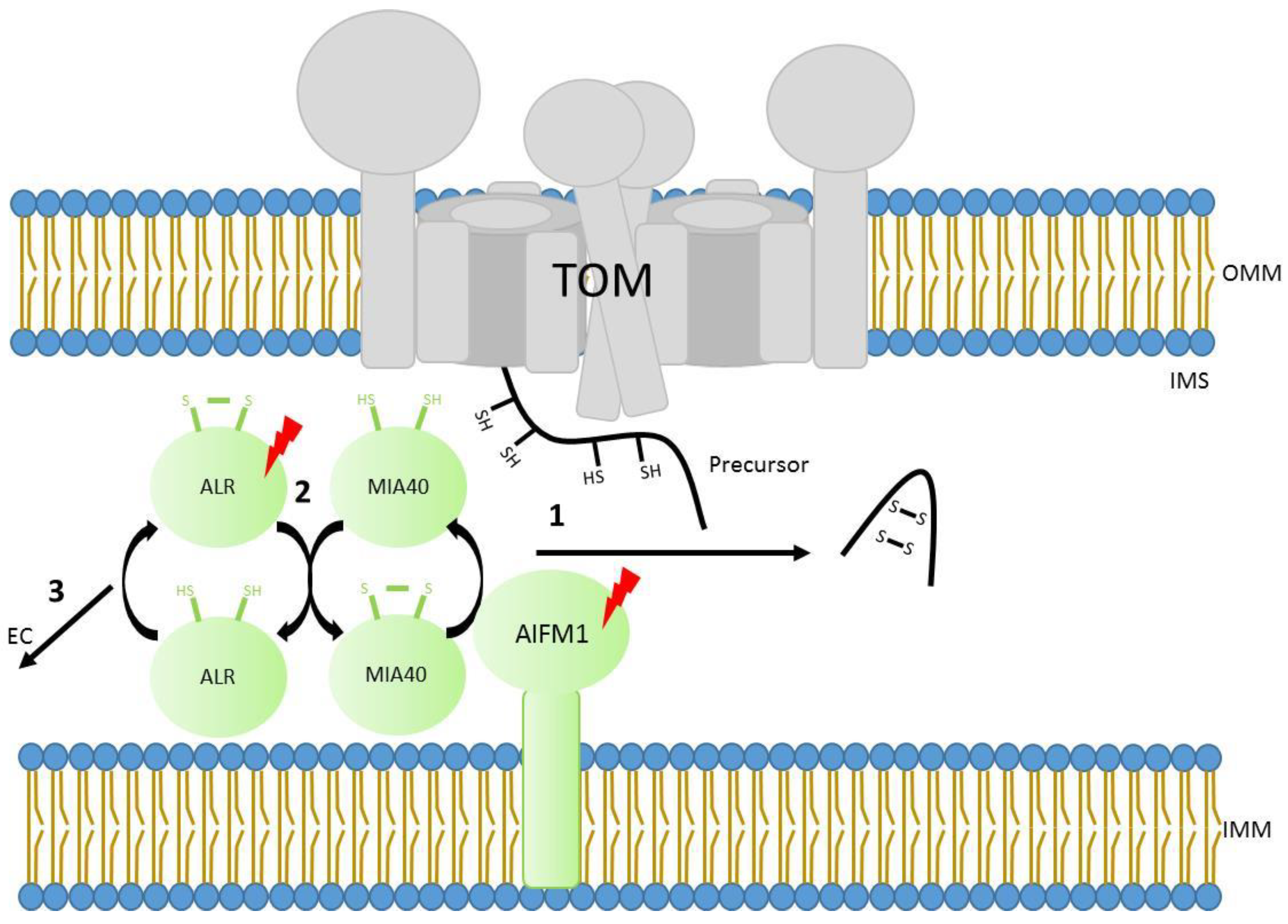
4. Import to the Intermembrane Space Using Disulfide Bridge Formation
5. Mutations in the Mitochondrial Disulfide Relay System
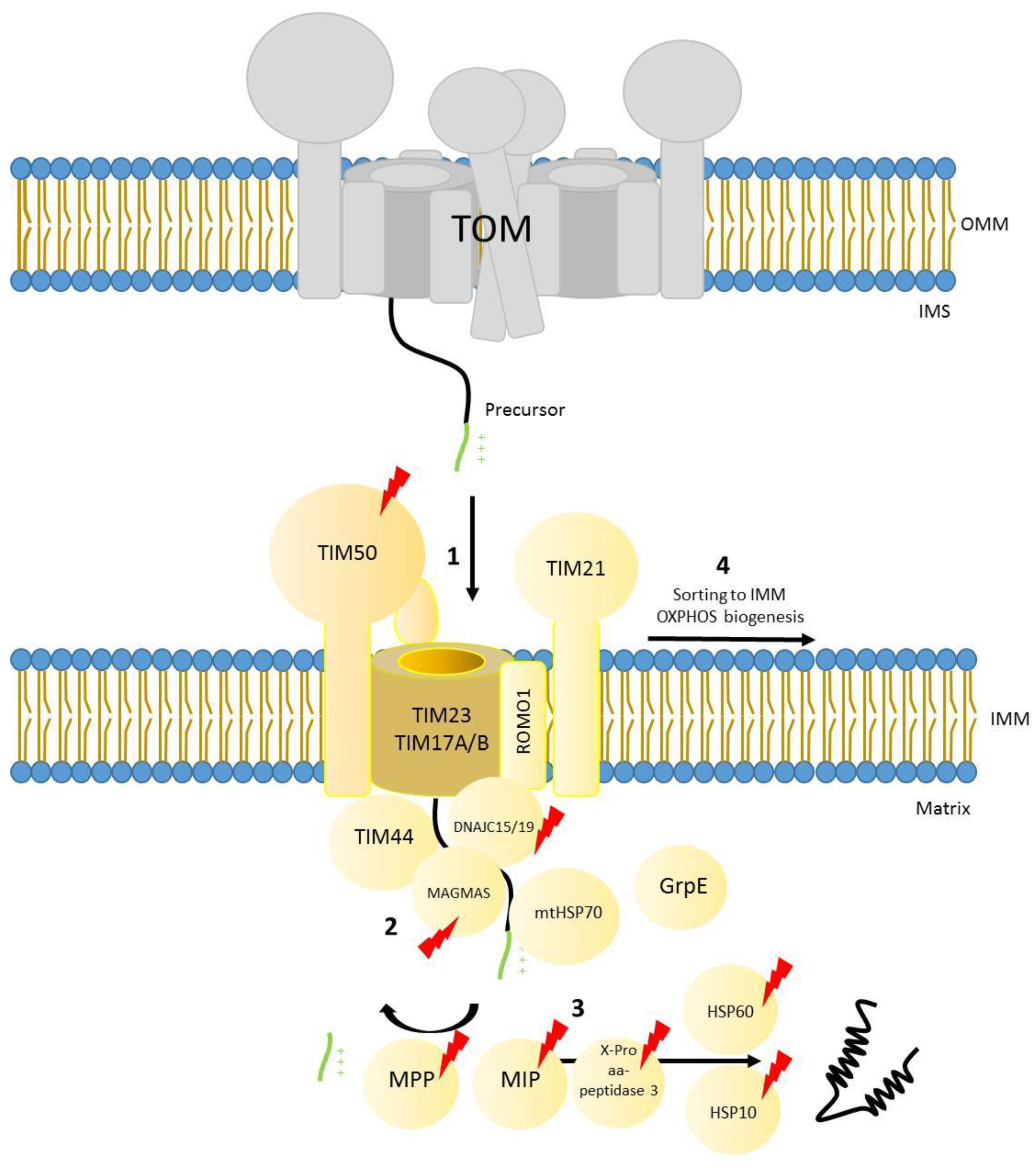
6. The Presequence Pathway
7. Defects in Presequence Dependent Import
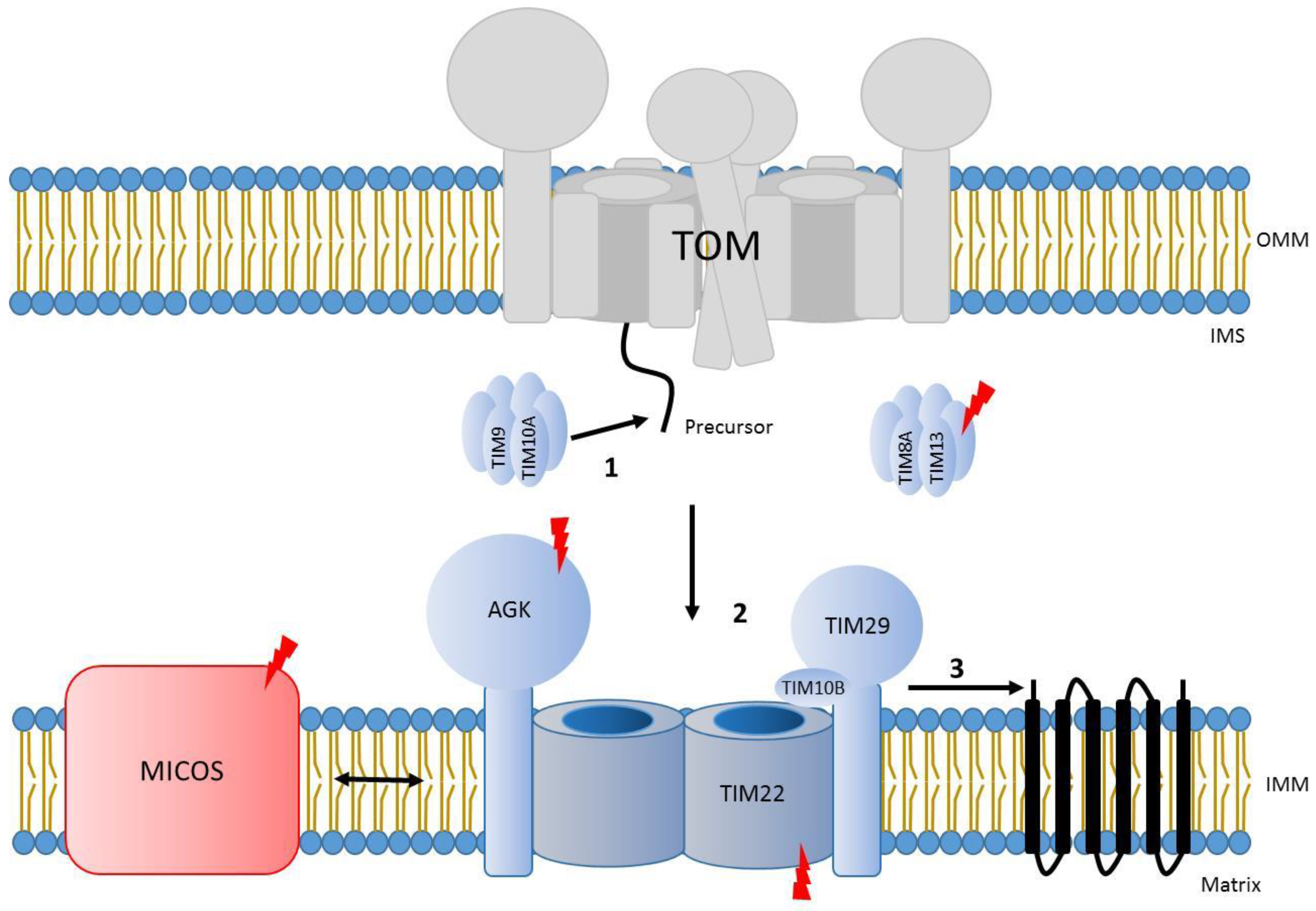
8. Carrier Translocase Mediated Import across the Inner Membrane
9. Defects in Carrier Transport Associated with Disease
10. Other Import Pathways
11. Expanding the Genetic Landscape of Mitochondrial Protein Import Disorders
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Hoess, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [Green Version]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Fielden, L.F.; Stojanovski, D. Mitochondrial protein transport in health and disease. Semin. Cell Dev. Biol. 2018, 76, 142–153. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Callegari, S.; Cruz-Zaragoza, L.D.; Rehling, P. From TOM to the TIM23 complex—Handing over of a precursor. Biol. Chem. 2020, 401, 709–721. [Google Scholar] [CrossRef]
- Priesnitz, C.; Pfanner, N.; Becker, T. Studying protein import into mitochondria. Methods Cell Biol. 2020, 155, 45–79. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Ferrari, A.; Del’Olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef] [PubMed]
- Van Wilpe, S.; Ryan, M.T.; Hill, K.; Maarse, A.C.; Meisinger, C.; Brix, J.; Dekker, P.J.T.; Moczko, M.; Wagner, R.; Meijer, M.; et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nat. Cell Biol. 1999, 401, 485–489. [Google Scholar] [CrossRef]
- Abe, Y.; Shodai, T.; Muto, T.; Mihara, K.; Torii, H.; Nishikawa, S.-I.; Endo, T.; Kohda, D. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell 2000, 100, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Fukui, K.; Takahashi, H.; Kitamura, S.; Shiota, T.; Terao, K.; Uchida, M.; Esaki, M.; Nishikawa, S.-I.; Yoshihisa, T.; et al. Roles of Tom70 in Import of Presequence-containing Mitochondrial Proteins. J. Biol. Chem. 2009, 284, 31635–31646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backes, S.; Hess, S.; Boos, F.; Woellhaf, M.W.; Gödel, S.; Jung, M.; Mühlhaus, T.; Herrmann, J.M. Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J. Cell Biol. 2018, 217, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, K.; Model, K.; Ryan, M.T.; Dietmeier, K.; Martin, F.; Wagner, R.; Pfanner, N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nat. Cell Biol. 1998, 395, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Melin, J.; Schulz, C.; Wrobel, L.; Bernhard, O.; Chacinska, A.; Jahn, O.; Schmidt, B.; Rehling, P. Presequence Recognition by the Tom40 Channel Contributes to Precursor Translocation into the Mitochondrial Matrix. Mol. Cell. Biol. 2014, 34, 3473–3485. [Google Scholar] [CrossRef] [Green Version]
- Kuszak, A.; Jacobs, D.; Gurnev, P.A.; Shiota, T.; Louis, J.M.; Lithgow, T.; Bezrukov, S.M.; Rostovtseva, T.K.; Buchanan, S.K. Evidence of Distinct Channel Conformations and Substrate Binding Affinities for the Mitochondrial Outer Membrane Protein Translocase Pore Tom40. J. Biol. Chem. 2015, 290, 26204–26217. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Mihara, K. Identification of Tom5 and Tom6 in the preprotein translocase complex of human mitochondrial outer membrane. Biochem. Biophys. Res. Commun. 2008, 369, 958–963. [Google Scholar] [CrossRef]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kühlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, X.; Zhang, L.; Yi, J.; Ma, Q.; Yin, J.; Zhuo, W.; Gu, J.; Yang, M. Atomic structure of human TOM core complex. Cell Discov. 2020, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.; Park, E. Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2019, 26, 1158–1166. [Google Scholar] [CrossRef]
- Araiso, Y.; Tsutsumi, A.; Qiu, J.; Imai, K.; Shiota, T.; Song, J.; Lindau, C.; Wenz, L.-S.; Sakaue, H.; Yunoki, K.; et al. Structure of the mitochondrial import gate reveals distinct preprotein paths. Nat. Cell Biol. 2019, 575, 395–401. [Google Scholar] [CrossRef]
- Bolliger, L.; Junne, T.; Schatz, G.; Lithgow, T. Acidic receptor domains on both sides of the outer membrane mediate translocation of precursor proteins into yeast mitochondria. EMBO J. 1995, 14, 6318–6326. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, T.; Nishikawa, S.-I.; Shin, I.; Schultz, P.G.; Endo, T. Probing the environment along the protein import pathways in yeast mitochondria by site-specific photocrosslinking. Proc. Natl. Acad. Sci. USA 1997, 94, 485–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokranjac, D.; Sichting, M.; Popov-Čeleketić, D.; Mapa, K.; Gevorkyan-Airapetov, L.; Zohary, K.; Hell, K.; Azem, A.; Neupert, W. Role of Tim50 in the Transfer of Precursor Proteins from the Outer to the Inner Membrane of Mitochondria. Mol. Biol. Cell 2009, 20, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.; Tan, K.; Shen, H.-H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular architecture of the active mitochondrial protein gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Esaki, M.; Kanamori, T.; Nishikawa, S.-I.; Shin, I.; Schultz, P.G.; Endo, T. Tom40 protein import channel binds to non-native proteins and prevents their aggregation. Nat. Struct. Mol. Biol. 2003, 10, 988–994. [Google Scholar] [CrossRef]
- Mahendran, K.R.; Romero-Ruiz, M.; Schlösinger, A.; Winterhalter, M.; Nussberger, S. Protein Translocation through Tom40: Kinetics of Peptide Release. Biophys. J. 2012, 102, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Romero-Ruiz, M.; Mahendran, K.R.; Eckert, R.; Winterhalter, M.; Nussberger, S. Interactions of Mitochondrial Presequence Peptides with the Mitochondrial Outer Membrane Preprotein Translocase TOM. Biophys. J. 2010, 99, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [Green Version]
- Gadir, N.; Haim-Vilmovsky, L.; Kraut-Cohen, J.; Gerst, J.E. Localization of mRNAs coding for mitochondrial proteins in the yeast Saccharomyces cerevisiae. RNA 2011, 17, 1551–1565. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Gold, V.A.; Chroscicki, P.; Bragoszewski, P.; Chacinska, A. Visualization of cytosolic ribosomes on the surface of mitochondria by electron cryo-tomography. EMBO Rep. 2017, 18, 1786–1800. [Google Scholar] [CrossRef]
- Lesnik, C.; Cohen, Y.; Atir-Lande, A.; Schuldiner, M.; Arava, Y. OM14 is a mitochondrial receptor for cytosolic ribosomes that supports co-translational import into mitochondria. Nat. Commun. 2014, 5, 5711. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins into Mitochondria. Protein J. 2019, 38, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Eliyahu, E.; Pnueli, L.; Melamed, D.; Scherrer, T.; Gerber, A.P.; Pines, O.; Rapaport, D.; Arava, Y. Tom20 mediates localization of mRNAs to mitochondria in a translation-dependent manner. Mol. Cell Biol. 2010, 30, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F. Molecular Chaperones Hsp90 and Hsp70 Deliver Preproteins to the Mitochondrial Import Receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Du, M.; Xie, J.; Luo, T.; Zhou, Y.; Zhang, K.; Li, J.; Chen, D.; Xu, P.; Jia, M.; et al. Mutations in TOMM70 lead to multi-OXPHOS deficiencies and cause severe anemia, lactic acidosis, and developmental delay. J. Hum. Genet. 2020, 65, 231–240. [Google Scholar] [CrossRef]
- Dutta, D.; Briere, L.C.; Kanca, O.; Marcogliese, P.C.; Walker, M.A.; High, F.A.; Vanderver, A.; Krier, J.; Carmichael, N.; Callahan, C.; et al. De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment. Hum. Mol. Genet. 2020, 29, 1568–1579. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Ronchi, D.; Lodi, T.; Fassone, E.; Tigano, M.; Lamperti, C.; Corti, S.; Bordoni, A.; Fortunato, F.; Nizzardo, M.; et al. The Mitochondrial Disulfide Relay System Protein GFER Is Mutated in Autosomal-Recessive Myopathy with Cataract and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2009, 84, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, L.; Holm, I.A.; Teot, L.A.; Anselm, I. Adrenal Insufficiency in Mitochondrial Disease. J. Child. Neurol. 2016, 31, 190–194. [Google Scholar] [CrossRef]
- North, K.; Korson, M.S.; Krawiecki, N.; Shoffner, J.M.; Holm, I.A. Oxidative phosphorylation defect associated with primary adrenal insufficiency. J. Pediatr. 1996, 128, 688–692. [Google Scholar] [CrossRef]
- Nambot, S.; Gavrilov, D.; Thevenon, J.; Bruel, A.; Bainbridge, M.; Rio, M.; Goizet, C.; Rotig, A.; Jaeken, J.; Niu, N.; et al. Further delineation of a rare recessive encephalomyopathy linked to mutations in GFER thanks to data sharing of whole exome sequencing data. Clin. Genet. 2017, 92, 188–198. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D’Adamo, P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-Linked Mitochondrial Encephalomyopathy Associated with a Mutation in Apoptosis-Inducing Factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Berger, I.; Ben-Neriah, Z.; Dor-Wolman, T.; Shaag, A.; Saada, A.; Zenvirt, S.; Raas-Rothschild, A.; Nadjari, M.; Kaestner, K.H.; Elpeleg, O. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol. Genet. Metab. 2011, 104, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Grunseich, C.; Sevrioukova, I.F.; Schindler, A.; Horkayne-Szakaly, I.; Lamperti, C.; Landouré, G.; Kennerson, M.L.; Burnett, B.G.; Bönnemann, C.; et al. Cowchock Syndrome Is Associated with a Mutation in Apoptosis-Inducing Factor. Am. J. Hum. Genet. 2012, 91, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, X.; Wang, J.; Liu, L.; Xie, Y.; Huang, S.; Pakhrin, P.S.; Jin, Q.; Zhu, C.; Tang, B.; et al. A novel AIFM1 mutation in a Chinese family with X-linked Charcot–Marie–Tooth disease type 4. Neuromuscul. Disord. 2018, 28, 652–659. [Google Scholar] [CrossRef]
- Ardissone, A.; Piscosquito, G.; Legati, A.; Langella, T.; Lamantea, E.; Garavaglia, B.; Salsano, E.; Farina, L.; Moroni, I.; Pareyson, D.; et al. A slowly progressive mitochondrial encephalomyopathy widens the spectrum of AIFM1 disorders. Neurol. 2015, 84, 2193–2195. [Google Scholar] [CrossRef] [Green Version]
- Kettwig, M.; Schubach, M.; Zimmermann, F.A.; Klinge, L.; Mayr, J.; Biskup, S.; Sperl, W.; Gärtner, J.; Huppke, P. From ventriculomegaly to severe muscular atrophy: Expansion of the clinical spectrum related to mutations in AIFM1. Mitochondrion 2015, 21, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Prabhu, S.P.; Lidov, H.G.; Shi, J.; Anselm, I.; Brownstein, C.A.; Bainbridge, M.N.; Beggs, A.; Vargas, S.O.; Agrawal, P.B. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Mol. Case Stud. 2017, 3, a001560. [Google Scholar] [CrossRef] [Green Version]
- Diodato, D.; Tasca, G.; Verrigni, D.; D’Amico, A.; Rizza, T.; Tozzi, G.; Martinelli, D.; Verardo, M.; Invernizzi, F.; Nasca, A.; et al. A novel AIFM1 mutation expands the phenotype to an infantile motor neuron disease. Eur. J. Hum. Genet. 2016, 24, 463–466. [Google Scholar] [CrossRef]
- Hu, B.; Wang, M.; Castoro, R.; Simmons, M.; Dortch, R.; Yawn, R.; Li, J. A novel missense mutation in AIFM1 results in axonal polyneuropathy and misassembly of OXPHOS complexes. Eur. J. Neurol. 2017, 24, 1499–1506. [Google Scholar] [CrossRef]
- Sancho, P.; Sánchez-Monteagudo, A.; Collado, A.; Marco-Marín, C.; Domínguez-González, C.; Camacho, A.; Knecht, E.; Espinós, C.; Lupo, V. A newly distal hereditary motor neuropathy caused by a rare AIFM1 mutation. Neurogenetics 2017, 18, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Guan, J.; Ealy, M.; Zhang, Q.; Wang, D.; Wang, H.; Zhao, Y.; Sheng, Z.; Campbell, C.A.; Wang, F.; et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J. Med. Genet. 2015, 52, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Mierzewska, H.; Rydzanicz, M.; Biegański, T.; Kosinska, J.; Mierzewska-Schmidt, M.; Ługowska, A.; Pollak, A.; Stawiński, P.; Walczak, A.; Kędra, A.; et al. Spondyloepimetaphyseal dysplasia with neurodegeneration associated withAIFM1mutation—A novel phenotype of the mitochondrial disease. Clin. Genet. 2017, 91, 30–37. [Google Scholar] [CrossRef]
- Miyake, N.; Wolf, N.I.; Cayami, F.K.; Crawford, J.; Bley, A.; Bulas, D.; Conant, A.; Bent, S.J.; Gripp, K.W.; Hahn, A.; et al. X-linked hypomyelination with spondylometaphyseal dysplasia (H-SMD) associated with mutations in AIFM1. Neurogenetics 2017, 18, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Heimer, G.; Eyal, E.; Zhu, X.; Ruzzo, E.; Marek-Yagel, D.; Sagiv, D.; Anikster, Y.; Reznik-Wolf, H.; Pras, E.; Levi, D.O.; et al. Mutations in AIFM1 cause an X-linked childhood cerebellar ataxia partially responsive to riboflavin. Eur. J. Paediatr. Neurol. 2018, 22, 93–101. [Google Scholar] [CrossRef]
- Kawarai, T.; Yamazaki, H.; Yamakami, K.; Tsukamoto-Miyashiro, A.; Kodama, M.; Rumore, R.; Caltagirone, C.; Nishino, I.; Orlacchio, A. A novel AIFM1 missense mutation in a Japanese patient with ataxic sensory neuronopathy and hearing impairment. J. Neurol. Sci. 2020, 409, 116584. [Google Scholar] [CrossRef]
- Pandolfo, M.; Rai, M.; Remiche, G.; Desmyter, L.; VanderNoot, I. Cerebellar ataxia, neuropathy, hearing loss, and intellectual disability due to AIFM1 mutation. Neurol. Genet. 2020, 6, e420. [Google Scholar] [CrossRef] [Green Version]
- Elrharchi, S.; Riahi, Z.; Salime, S.; Charoute, H.; Elkhattabi, L.; Boulouiz, R.; Kabine, M.; Bonnet, C.; Petit, C.; Barakat, A. Novel Mutation in AIFM1 Gene Associated with X-Linked Deafness in a Moroccan Family. Hum. Hered. 2021, 85, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shahrour, M.; Staretz-Chacham, O.; Dayan, D.; Stephen, J.; Weech, A.; Damseh, N.; Chen, H.P.; Edvardson, S.; Mazaheri, S.; Saada, A.; et al. Mitochondrial epileptic encephalopathy, 3-methylglutaconic aciduria and variable complex V deficiency associated with TIMM50 mutations. Clin. Genet. 2016, 91, 690–696. [Google Scholar] [CrossRef]
- Reyes, A.; Melchionda, L.; Burlina, A.; Robinson, A.J.; Ghezzi, D.; Zeviani, M. Mutations in TIMM50 compromise cell survival in OxPhos-dependent metabolic conditions. EMBO Mol. Med. 2018, 10, e8698. [Google Scholar] [CrossRef]
- Tort, F.; Ugarteburu, O.; Texidó, L.; Gea-Sorlí, S.; García-Villoria, J.; Ferrer-Cortès, X.; Arias, Á.; Matalonga, L.; Gort, L.; Ferrer, I.; et al. Mutations in TIMM50 cause severe mitochondrial dysfunction by targeting key aspects of mitochondrial physiology. Hum. Mutat. 2019, 40, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Mehawej, C.; Delahodde, A.; Legeai-Mallet, L.; Delague, V.; Kaci, N.; Desvignes, J.-P.; Kibar, Z.; Capo-Chichi, J.-M.; Chouery, E.; Munnich, A.; et al. The Impairment of MAGMAS Function in Human Is Responsible for a Severe Skeletal Dysplasia. PLoS Genet. 2014, 10, e1004311. [Google Scholar] [CrossRef]
- Moosa, S.; Fano, V.; Obregon, M.G.; Altmüller, J.; Thiele, H.; Nürnberg, P.; Nishimura, G.; Wollnik, B. A novel homozygousPAM16mutation in a patient with a milder phenotype and longer survival. Am. J. Med. Genet. Part A 2016, 170, 2436–2439. [Google Scholar] [CrossRef]
- Davey, K.M.; Parboosingh, J.S.; McLeod, D.R.; Chan, A.; Casey, R.A.; Ferreira, P.A.; Snyder, F.F.; Bridge, P.; Bernier, F.P. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 2005, 43, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Ojala, T.; Polinati, P.; Manninen, T.; Hiippala, A.; Rajantie, J.; Karikoski, R.; Suomalainen-Wartiovaara, A.; Tyni, T. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr. Res. 2012, 72, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Al Teneiji, A.; Siriwardena, K.; George, K.; Mital, S.; Mercimek-Mahmutoglu, S. Progressive cerebellar atrophy and a novel homozygous pathogenic DNAJC19 variant as a cause of dilated cardiomyopathy ataxia syndrome. Pediatr. Neurol. 2016, 62, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Ucar, S.K.; Mayr, J.; Feichtinger, R.G.; Canda, E.; Çoker, M.; Wortmann, S.B. Previously Unreported Biallelic Mutation in DNAJC19: Are Sensorineural Hearing Loss and Basal Ganglia Lesions Additional Features of Dilated Cardiomyopathy and Ataxia (DCMA) Syndrome? JIMD Rep. 2016, 35, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Luder, A.; et al. Mitochondrial Hsp60 Chaperonopathy Causes an Autosomal-Recessive Neurodegenerative Disorder Linked to Brain Hypomyelination and Leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusk, M.S.; Damgaard, B.; Risom, L.; Hansen, B.; Ostergaard, E. Hypomyelinating Leukodystrophy due to HSPD1 Mutations: A New Patient. Neuropediatrics 2016, 47, 332–335. [Google Scholar] [CrossRef]
- Hansen, J.J.; Dürr, A.; Cournu-Rebeix, I.; Georgopoulos, C.; Ang, D.; Nielsen, M.N.; Davoine, C.-S.; Brice, A.; Fontaine, B.; Gregersen, N.; et al. Hereditary Spastic Paraplegia SPG13 Is Associated with a Mutation in the Gene Encoding the Mitochondrial Chaperonin Hsp60. Am. J. Hum. Genet. 2002, 70, 1328–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, H.; Mittal, N.; Inomata, T.; Arimura, T.; Izumi, T.; Kimura, A.; Fukuda, K.; Makino, S. Dilated cardiomyopathy-linked heat shock protein family D member 1 mutations cause up-regulation of reactive oxygen species and autophagy through mitochondrial dysfunction. Cardiovasc. Res. 2021, 117, 1118–1131. [Google Scholar] [CrossRef]
- Bie, A.S.; Fernandez-Guerra, P.; Birkler, R.I.D.; Nisemblat, S.; Pelnena, D.; Lu, X.; Deignan, J.L.; Lee, H.; Dorrani, N.; Corydon, T.J.; et al. Effects of a Mutation in the HSPE1 Gene Encoding the Mitochondrial Co-chaperonin HSP10 and Its Potential Association with a Neurological and Developmental Disorder. Front. Mol. Biosci. 2016, 3, 65. [Google Scholar] [CrossRef]
- Jobling, R.K.; Assoum, M.; Gakh, O.; Blaser, S.; Raiman, J.A.; Mignot, C.; Roze, E.; Durr, A.; Brice, A.; Lévy, N.; et al. PMPCAmutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 2015, 138, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Choquet, K.; Zurita-Rendón, O.; La Piana, R.; Yang, S.; Dicaire, M.-J.; Boycott, K.M.; Majewski, J.; Shoubridge, E.A.; Brais, B.; Tétreault, M.; et al. Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain 2015, 139. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.; Anselm, I.; Shi, J.; Bale, T.A.; Towne, M.; Schmitz-Abe, K.; Crowley, L.; Giani, F.C.; Kazerounian, S.; Markianos, K.; et al. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-α protein (PMPCA) cause a severe mitochondrial disease. Mol. Case Stud. 2016, 2, a000786. [Google Scholar] [CrossRef] [Green Version]
- Vögtle, F.-N.; Brändl, B.; Larson, A.; Pendziwiat, M.; Friederich, M.W.; White, S.M.; Basinger, A.; Kücükköse, C.; Muhle, H.; Jähn, J.A.; et al. Mutations in PMPCB Encoding the Catalytic Subunit of the Mitochondrial Presequence Protease Cause Neurodegeneration in Early Childhood. Am. J. Hum. Genet. 2018, 102, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Eldomery, M.K.; Akdemir, Z.C.; Vögtle, F.-N.; Charng, W.-L.; Mulica, P.; Rosenfeld, J.A.; Gambin, T.; Gu, S.; Burrage, L.C.; Al Shamsi, A.; et al. MIPEP recessive variants cause a syndrome of left ventricular non-compaction, hypotonia, and infantile death. Genome Med. 2016, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Otto, E.A.; Ramaswami, G.; Janssen, S.; Chaki, M.; Allen, S.J.; Zhou, W.; Airik, R.; Hurd, T.W.; Ghosh, A.K.; Wolf, M.T.; et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J. Med. Genet. 2010, 48, 105–116. [Google Scholar] [CrossRef]
- O’Toole, J.F.; Liu, Y.; Davis, E.; Westlake, C.J.; Attanasio, M.; Otto, E.A.; Seelow, D.; Nurnberg, G.; Becker, C.; Nuutinen, M.; et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J. Clin. Investig. 2010, 120, 791–802. [Google Scholar] [CrossRef]
- Alizadeh, R.; Jamshidi, S.; Keramatipour, M.; Moeinian, P.; Hosseini, R.; Otukesh, H.; Talebi, S.; Hospital, T.A.A.C. Whole Exome Sequencing Reveals a XPNPEP3 Novel Mutation Causing Nephronophthisis in a Pediatric Patient. Iran. Biomed. J. 2020, 24, 400–403. [Google Scholar] [CrossRef]
- Jin, H.; May, M.; Tranebjærg, L.; Kendall, E.; Fontán, G.; Jackson, J.; Subramony, S.; Arena, F.; Lubs, H.; Smith, S.; et al. A novel X–linked gene, DDP, shows mutations in families with deafness (DFN–1), dystonia, mental deficiency and blindness. Nat. Genet. 1996, 14, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Wooten, G.F. A novel deafness/dystonia peptide gene mutation that causes dystonia in female carriers of Mohr-Tranebjaerg syndrome. Ann. Neurol. 2001, 50, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.A.; Del Castillo, I.; Macaya, A.; Medá, C.; Villamar, M.; Moreno-Pelayo, M.A.; Moreno, F. A novel mutation in the gene encoding TIMM8a, a component of the mitochondrial protein translocase complexes, in a Spanish familial case of deafness-dystonia (Mohr–Tranebjaerg) syndrome. Am. J. Med. Genet. Part. A 2006, 140, 392–397. [Google Scholar] [CrossRef]
- Tranebjaerg, L.; Van Ghelue, M.; Nilssen, O.; Hodes, M.E.; Dlouhy, S.R.; Farlow, M.R.; Hamel, B.; Arts, W.F.M.; Jankovic, J.; Beach, J.; et al. Jensen syndrome is allelic to Mohr–Tranebjaerg syndrome and both are caused by stop mutations in the DDP gene. Am. J. Hum. Genet. 1997, 61S, A349, [abstract]. (accessed on 28 June 2021). [Google Scholar]
- Ujike, H.; Tanabe, Y.; Takehisa, Y.; Hayabara, T.; Kuroda, S. A Family With X-linked Dystonia-Deafness Syndrome with a Novel Mutation of the DDP Gene. Arch. Neurol. 2001, 58, 1004–1007. [Google Scholar] [CrossRef]
- Blesa, J.R.; Solano, A.; Briones, P.; Prieto-Ruiz, J.A.; Hernández-Yago, J.; Coria, F. Molecular Genetics of a Patient with Mohr–Tranebjaerg Syndrome due to a New Mutation in the DDP1 Gene. NeuroMolecular Med. 2007, 9, 285–291. [Google Scholar] [CrossRef]
- Song, P.; Guan, Y.; Chen, X.; Wu, C.; Qiao, A.; Jiang, H.; Li, Q.; Huang, Y.; Huang, W.; Xu, M.; et al. Frameshift mutation of Timm8a1 gene in mouse leads to an abnormal mitochondrial structure in the brain, correlating with hearing and memory impairment. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Tranebjærg, L.; Hamel, B.C.; Gabreels, F.J.; O Renier, W.; Van Ghelue, M. A de novo missense mutation in a critical domain of the X-linked DDP gene causes the typical deafness–dystonia–optic atrophy syndrome. Eur. J. Hum. Genet. 2000, 8, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Binder, J.; Hofmann, S.; Kreisel, S.; Wöhrle, J.C.; Bäzner, H.; Krauss, J.K.; Hennerici, M.G.; Bauer, M.F. Clinical and molecular findings in a patient with a novel mutation in the deafness-dystonia peptide (DDP1) gene. Brain 2003, 126, 1814–1820. [Google Scholar] [CrossRef] [Green Version]
- Ezquerra, M.; Campdelacreu, J.; Muñoz, E.; Tolosa, E.; Martí, M.J. A Novel Intronic Mutation in the DDP1 Gene in a Family With X-linked Dystonia-Deafness Syndrome. Arch. Neurol. 2005, 62, 306–308. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.T.; Edwards, M.J.; Tyson, J.; Quinn, N.P.; Bitner-Glindzicz, M.; Bhatia, K.P. Blepharospasm and limb dystonia caused by Mohr-Tranebjaerg syndrome with a novel splice-site mutation in the deafness/dystonia peptide gene. Mov. Disord. 2007, 22, 1328–1331. [Google Scholar] [CrossRef]
- Aguirre, L.A.; Pérez-Bas, M.; Villamar, M.; López-Ariztegui, M.A.; Moreno-Pelayo, M.A.; Moreno, F.; Del Castillo, I. A Spanish sporadic case of deafness–dystonia (Mohr-Tranebjaerg) syndrome with a novel mutation in the gene encoding TIMM8a, a component of the mitochondrial protein translocase complexes. Neuromuscul. Disord. 2008, 18, 979–981. [Google Scholar] [CrossRef]
- Pizzuti, A.; Fabbrini, G.; Salehi, L.; Vacca, L.; Inghilleri, M.; Dallapiccola, B.; Berardelli, A. Focal dystonia caused by Mohr-Tranebjaerg syndrome with complete deletion of the DDP1 gene. Neurology 2004, 62, 1021–1022. [Google Scholar] [CrossRef]
- Richter, D.; Conley, M.E.; Rohrer, J.; Myers, L.A.; Zahradka, K.; Ic, J.K.; Sertic, J.; Rukavina, A.S. A contiguous deletion syndrome of X-linked agammaglobulinemia and sensorineural deafness. Pediatr. Allergy Immunol. 2001, 12, 107–111. [Google Scholar] [CrossRef]
- Šedivá, A.; Smith, C.I.E.; Asplund, A.C.; Hadač, J.; Janda, A.; Zeman, J.; Hansíková, H.; Dvořáková, L.; Mrázová, L.; Velbri, S.; et al. Contiguous X-chromosome Deletion Syndrome Encompassing the BTK, TIMM8A, TAF7L, and DRP2 Genes. J. Clin. Immunol. 2007, 27, 640–646. [Google Scholar] [CrossRef]
- Arai, T.; Zhao, M.; Kanegane, H.; Van Zelm, M.C.; Futatani, T.; Yamada, M.; Ariga, T.; Ochs, H.D.; Miyawaki, T.; Oh-Ishi, T. Genetic analysis of contiguous X-chromosome deletion syndrome encompassing the BTK and TIMM8A genes. J. Hum. Genet. 2011, 56, 577–582. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.; Hershman, S.; Lim, S.C.; Lieber, D.S.; Tucker, E.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, K.; MacKay, N.; Levandovskiy, V.; Blaser, S.; Raiman, J.; Kantor, P.; Ackerley, C.; Robinson, B.H.; Schulze, A.; Cameron, J.M. Mitochondrial citrate synthase crystals: Novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations. Mol. Genet. Metab. 2013, 108, 40–50. [Google Scholar] [CrossRef]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; Bashir, R.A.; Ahting, U.; Feichtinger, R.G.; Mayr, J.A.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allali, S.; Dorboz, I.; Samaan, S.; Slama, A.; Rambaud, C.; Boespflug-Tanguy, O.; Sarret, C. Mutation in the AGK gene in two siblings with unusual Sengers syndrome. Metab. Brain Dis. 2017, 32, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Guleray, N.; Kosukcu, C.; Taskiran, Z.E.; Simsek Kiper, P.O.; Utine, G.E.; Gucer, S.; Tokatli, A.; Boduroglu, K.; Alikasifoglu, M. Atypical Presentation of Sengers Syndrome: A Novel Mutation Revealed with Postmortem Genetic Testing. Fetal Pediatr. Pathol. 2019, 39, 163–171. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Khan, A.O.; Mohamed, J.Y.; Alghamdi, M.H.; Alkuraya, F.S. Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus. Hum. Mutat. 2012, 33, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Callegari, S.; Emperador, S.; Thompson, K.; Aich, A.; E Topol, S.; Spencer, E.G.; McFarland, R.; Ruiz-Pesini, E.; Torkamani, A.; et al. Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy. Hum. Mol. Genet. 2018, 27, 4135–4144. [Google Scholar] [CrossRef]
- Guarani, V.; Jardel, C.; Chrétien, D.; Lombès, A.; Bénit, P.; Labasse, C.; Lacène, E.; Bourillon, A.; Imbard, A.; Benoist, J.-F.; et al. QIL1 mutation causes MICOS disassembly and early onset fatal mitochondrial encephalopathy with liver disease. eLife 2016, 5. [Google Scholar] [CrossRef]
- Zeharia, A.; Friedman, J.; Tobar, A.; Saada, A.; Konen, O.; Fellig, Y.; Shaag, A.; Nunnari, J.; Elpeleg, O. Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19orf70), a MICOS complex subunit. Eur. J. Hum. Genet. 2016, 24, 1778–1782. [Google Scholar] [CrossRef] [Green Version]
- Kishita, Y.; Shimura, M.; Kohda, M.; Akita, M.; Imai-Okazaki, A.; Yatsuka, Y.; Nakajima, Y.; Ito, T.; Ohtake, A.; Murayama, K.; et al. A novel homozygous variant in MICOS13/QIL1 causes hepato-encephalopathy with mitochondrial DNA depletion syndrome. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef]
- Gödiker, J.; Grüneberg, M.; Duchesne, I.; Reunert, J.; Rust, S.; Westermann, C.; Wada, Y.; Classen, G.; Langhans, C.D.; Schlingmann, K.P.; et al. QIL1-dependent assembly of MICOS complex–lethal mutation in C19ORF70 resulting in liver disease and severe neurological retardation. J. Hum. Genet. 2018, 63, 707–716. [Google Scholar] [CrossRef]
- Benincá, C.; Zanette, V.; Brischigliaro, M.; Johnson, M.; Reyes, A.; Valle, D.A.D.; Robinson, A.J.; Degiorgi, A.; Yeates, A.; Telles, B.A.; et al. Mutation in the MICOS subunit gene APOO (MIC26) associated with an X-linked recessive mitochondrial myopathy, lactic acidosis, cognitive impairment and autistic features. J. Med. Genet. 2021, 58, 155–167. [Google Scholar] [CrossRef]
- Elouej, S.; Harhouri, K.; Le Mao, M.; Baujat, G.; Nampoothiri, S.; Kayserili, H.; Al Menabawy, N.; Selim, L.; Paneque, A.L.; Kubisch, C.; et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuán Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004, 23, 3735–3746. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.M.; Milenkovic, D.; Guiard, B.; Pfanner, N.; Chacinska, A. Precursor Oxidation by Mia40 and Erv1 Promotes Vectorial Transport of Proteins into the Mitochondrial Intermembrane Space. Mol. Biol. Cell 2008, 19, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztolsztener, M.E.; Brewinska, A.; Guiard, B.; Chacinska, A. Disulfide Bond Formation: Sulfhydryl Oxidase ALR Controls Mitochondrial Biogenesis of Human MIA40. Traffic 2013, 14, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Peleh, V.; Cordat, E.; Herrmann, J.M. Mia40 is a trans-site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. eLife 2016, 5, e16177. [Google Scholar] [CrossRef] [PubMed]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A Disulfide Relay System in the Intermembrane Space of Mitochondria that Mediates Protein Import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Cefaro, C.; Baffoni, S.C.; Gallo, A.; Martinelli, M.; Sideris, D.P.; Katrakili, N.; Tokatlidis, K. MIA40 is an oxidoreductase that catalyzes oxidative protein folding in mitochondria. Nat. Struct. Mol. Biol. 2009, 16, 198–206. [Google Scholar] [CrossRef]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Küttner, V.; Bien, M.; Kauff, F.; et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol. Biol. Cell 2013, 24, 2160–2170. [Google Scholar] [CrossRef]
- Meyer, K.; Buettner, S.; Ghezzi, D.; Zeviani, M.; Bano, D.; Nicotera, P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015, 6, e1814. [Google Scholar] [CrossRef] [PubMed]
- Hangen, E.; Féraud, O.; Lachkar, S.; Mou, H.; Doti, N.; Fimia, G.M.; Lam, N.-V.; Zhu, C.; Godin, I.; Muller, K.; et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol. Cell 2015, 58, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Vahsen, N.; Candé, C.; Brière, J.-J.; Bénit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; O Pequignot, M.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Joza, N.; Oudit, G.Y.; Brown, D.; Bénit, P.; Kassiri, Z.; Vahsen, N.; Benoit, L.; Patel, M.M.; Nowikovsky, K.; Vassault, A.; et al. Muscle-Specific Loss of Apoptosis-Inducing Factor Leads to Mitochondrial Dysfunction, Skeletal Muscle Atrophy, and Dilated Cardiomyopathy. Mol. Cell. Biol. 2005, 25, 10261–10272. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Yu, B.; Joza, N.; Bénit, P.; Meneses, J.; Firpo, M.; Rustin, P.; Penninger, J.M.; Martin, G.R. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc. Natl. Acad. Sci. USA 2006, 103, 9918–9923. [Google Scholar] [CrossRef] [Green Version]
- Modjtahedi, N.; Tokatlidis, K.; Dessen, P.; Kroemer, G. Mitochondrial Proteins Containing Coiled-Coil-Helix-Coiled-Coil-Helix (CHCH) Domains in Health and Disease. Trends Biochem. Sci. 2016, 41, 245–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, G. Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. BioSyst. 2010, 6, 2459–2470. [Google Scholar] [CrossRef] [PubMed]
- Petrungaro, C.; Zimmermann, K.M.; Küttner, V.; Fischer, M.; Dengjel, J.; Bogeski, I.; Riemer, J. The Ca2+-Dependent Release of the Mia40-Induced MICU1-MICU2 Dimer from MCU Regulates Mitochondrial Ca2+ Uptake. Cell Metab. 2015, 22, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R.; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunitNDUFB10result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2016, 26, 702–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bano, D.; Prehn, J.H. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevrioukova, I.F. Structure/Function Relations in AIFM1 Variants Associated with Neurodegenerative Disorders. J. Mol. Biol. 2016, 428, 3650–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischhof, L.; Gioran, A.; Sonntag-Bensch, D.; Piazzesi, A.; Stork, M.; Nicotera, P.; Bano, D. A disease-associated Aifm1 variant induces severe myopathy in knockin mice. Mol. Metab. 2018, 13, 10–23. [Google Scholar] [CrossRef]
- Troulinaki, K.; Büttner, S.; Cots, A.M.; Maida, S.; Meyer, K.; Bertan, F.; Gioran, A.; Piazzesi, A.; Fornarelli, A.; Nicotera, P.; et al. WAH-1/AIF regulates mitochondrial oxidative phosphorylation in the nematode Caenorhabditis elegans. Cell Death Discov. 2018, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global Analysis of the Mitochondrial N-Proteome Identifies a Processing Peptidase Critical for Protein Stability. Cell 2009, 139, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Vögtle, F.-N.; Prinz, C.; Kellermann, J.; Lottspeich, F.; Pfanner, N.; Meisinger, C. Mitochondrial protein turnover: Role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol. Biol. Cell 2011, 22, 2135–2143. [Google Scholar] [CrossRef]
- Veling, M.T.; Reidenbach, A.G.; Freiberger, E.C.; Kwiecien, N.W.; Hutchins, P.D.; Drahnak, M.J.; Jochem, A.; Ulbrich, A.; Rush, M.J.; Russell, J.D.; et al. Multi-omic Mitoprotease Profiling Defines a Role for Oct1p in Coenzyme Q Production. Mol. Cell 2017, 68, 970–977.e11. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, J.; Horwich, A.L.; Neupert, W.; Hartl, F.-U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nat. Cell Biol. 1989, 341, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höhfeld, J.; Hartl, F.U. Role of the chaperonin cofactor Hsp10 in protein folding and sorting in yeast mitochondria. J. Cell Biol. 1994, 126, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.F.; Gempel, K.; Reichert, A.; Rappold, G.A.; Lichtner, P.; Gerbitz, K.-D.; Neupert, W.; Brunner, M.; Hofmann, S. Genetic and structural characterization of the human mitochondrial inner membrane translocase. J. Mol. Biol. 1999, 289, 69–82. [Google Scholar] [CrossRef]
- Guo, Y.; Cheong, N.; Zhang, Z.; De Rose, R.; Deng, Y.; Farber, S.A.; Fernandes-Alnemri, T.; Alnemri, E.S. Tim50, a Component of the Mitochondrial Translocator, Regulates Mitochondrial Integrity and Cell Death. J. Biol. Chem. 2004, 279, 24813–24825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Srivastava, S.; Krishna, L.; D’Silva, P. Unraveling the Intricate Organization of Mammalian Mitochondrial Presequence Translocases: Existence of Multiple Translocases for Maintenance of Mitochondrial Function. Mol. Cell. Biol. 2014, 34, 1757–1775. [Google Scholar] [CrossRef] [Green Version]
- Truscott, K.; Kovermann, P.; Geissler, A.; Merlin, A.; Meijer, M.; Driessen, A.J.; Rassow, J.; Pfanner, N.; Wagner, R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Genet. 2001, 8, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Ruan, M.; Li, Y.; Yang, J.; Bai, S.; Richter, C.; Schwalbe, H.; Xie, C.; Shen, B.; Wang, J. Solution structure of the voltage-gated Tim23 channel in complex with a mitochondrial presequence peptide. Cell Res. 2020, 1–4. [Google Scholar] [CrossRef]
- Bauer, M.F.; Sirrenberg, C.; Neupert, W.; Brunner, M. Role of Tim23 as Voltage Sensor and Presequence Receptor in Protein Import into Mitochondria. Cell 1996, 87, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Meinecke, M. Tim50 Maintains the Permeability Barrier of the Mitochondrial Inner Membrane. Science 2006, 312, 1523–1526. [Google Scholar] [CrossRef]
- Martin, J.; Mahlke, K.; Pfanner, N. Role of an energized inner membrane in mitochondrial protein import. Delta psi drives the movement of presequences. J. Biol. Chem. 1991, 266, 18051–18057. [Google Scholar] [CrossRef]
- Denkert, N.; Schendzielorz, A.B.; Barbot, M.; Versemann, L.; Richter, F.; Rehling, P.; Meinecke, M. Cation selectivity of the presequence translocase channel Tim23 is crucial for efficient protein import. eLife 2017, 6, e28324. [Google Scholar] [CrossRef]
- Bhattacharyya, T.; Karnezis, A.N.; Murphy, S.P.; Hoang, T.; Freeman, B.C.; Phillips, B.; Morimoto, R.I. Cloning and Subcellular Localization of Human Mitochondrial hsp70. J. Biol. Chem. 1995, 270, 1705–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, J.; Fischer, H.; Craievich, A.; Hansen, L.D.; Ramos, C. Free Human Mitochondrial GrpE Is a Symmetric Dimer in Solution. J. Biol. Chem. 2003, 278, 35337–35344. [Google Scholar] [CrossRef] [Green Version]
- Chacinska, A.; Lind, M.; Frazier, A.; Dudek, J.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; Truscott, K.N.; Guiard, B.; et al. Mitochondrial Presequence Translocase: Switching between TOM Tethering and Motor Recruitment Involves Tim21 and Tim17. Cell 2005, 120, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Joshi, N.; Chittoor, B.; Samji, P.; D’Silva, P. Role of Magmas in protein transport and human mitochondria biogenesis. Hum. Mol. Genet. 2010, 19, 1248–1262. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC Links Mitochondrial Protein Translocation to Respiratory-Chain Assembly and Translational Regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Srivastava, S.; D’Silva, P. Functional Diversity of Human Mitochondrial J-proteins Is Independent of Their Association with the Inner Membrane Presequence Translocase. J. Biol. Chem. 2016, 291, 17345–17359. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Dennerlein, S.; Nikolov, M.; Jans, D.C.; Naumenko, N.; Aich, A.; MacVicar, T.; Linden, A.; Jakobs, S.; Urlaub, H.; et al. ROMO1 is a constituent of the human presequence translocase required for YME1L protease import. J. Cell Biol. 2019, 218, 598–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennerlein, S.; Rehling, P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 2015, 128, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsör, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483.e10. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-Regulated Translational Attenuation Adapts Mitochondrial Protein Import through Tim17A Degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef] [Green Version]
- MacVicar, T.; Ohba, Y.; Nolte, H.; Mayer, F.C.; Tatsuta, T.; Sprenger, H.-G.; Lindner, B.; Zhao, Y.; Li, J.; Bruns, C.; et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 2019, 575, 361–365. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.; Langer, T. DNAJC19, a Mitochondrial Cochaperone Associated with Cardiomyopathy, Forms a Complex with Prohibitins to Regulate Cardiolipin Remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Rehling, P.; Brandner, K.; Pfanner, N. Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol. 2004, 5, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Neupert, W. Distinct steps in the import of ADP/ATP carrier into mitochondria. J. Biol. Chem. 1987, 262, 7528–7536. [Google Scholar] [CrossRef]
- Zara, V.; Palmisano, I.; Rassow, J.; Palmieri, F. Biogenesis of the dicarboxylate carrier (DIC): Translocation across the mitochondrial outer membrane and subsequent release from the TOM channel are membrane potential-independent. J. Mol. Biol. 2001, 310, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.; Damoiseaux, R.; Glavin, J.D.; Dabir, D.V.; Walker, S.S.; Koehler, C.M. Substrate specificity of the TIM22 mitochondrial import pathway revealed with small molecule inhibitor of protein translocation. Proc. Natl. Acad. Sci. USA 2010, 107, 9578–9583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callegari, S.; Richter, F.; Chojnacka, K.; Jans, D.C.; Lorenzi, I.; Pacheu-Grau, D.; Jakobs, S.; Lenz, C.; Urlaub, H.; Dudek, J.; et al. TIM29 is a subunit of the human carrier translocase required for protein transport. FEBS Lett. 2016, 590, 4147–4158. [Google Scholar] [CrossRef]
- Kang, Y.; Baker, M.J.; Liem, M.; Louber, J.; McKenzie, M.; Atukorala, I.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S.; Stojanovski, D. Tim29 is a novel subunit of the human TIM22 translocase and is involved in complex assembly and stability. eLife 2016, 5, e17463. [Google Scholar] [CrossRef]
- Kang, Y.; Stroud, D.; Baker, M.J.; DE Souza, D.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e5. [Google Scholar] [CrossRef]
- Vukotic, M.; Nolte, H.; König, T.; Saita, S.; Ananjew, M.; Krüger, M.; Tatsuta, T.; Langer, T. Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 2017, 67, 471–483.e7. [Google Scholar] [CrossRef]
- Gomkale, R.; Cruz-Zaragoza, L.D.; Suppanz, I.; Guiard, B.; Montoya, J.; Callegari, S.; Pacheu-Grau, D.; Warscheid, B.; Rehling, P. Defining the Substrate Spectrum of the TIM22 Complex Identifies Pyruvate Carrier Subunits as Unconventional Cargos. Curr. Biol. 2020, 30, 1119–1127.e5. [Google Scholar] [CrossRef] [PubMed]
- Rampelt, H.; Sucec, I.; Bersch, B.; Horten, P.; Perschil, I.; Martinou, J.-C.; Van Der Laan, M.; Wiedemann, N.; Schanda, P.; Pfanner, N. The mitochondrial carrier pathway transports non-canonical substrates with an odd number of transmembrane segments. BMC Biol. 2020, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.D.; Hock, D.H.; Fujihara, K.M.; Palmer, C.S.; Frazier, A.E.; Low, Y.C.; Kang, Y.; Ang, C.-S.; Clemons, N.J.; Thorburn, D.R.; et al. The TIM22 complex mediates the import of sideroflexins and is required for efficient mitochondrial one-carbon metabolism. Mol. Biol. Cell 2021, 32, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Beverly, K.N.; Sawaya, M.R.; Schmid, E.; Koehler, C.M. The Tim8–Tim13 Complex Has Multiple Substrate Binding Sites and Binds Cooperatively to Tim23. J. Mol. Biol. 2008, 382, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Gebert, N.; Chacinska, A.; Wagner, K.; Guiard, B.; Koehler, C.M.; Rehling, P.; Pfanner, N.; Wiedemann, N. Assembly of the three small Tim proteins precedes docking to the mitochondrial carrier translocase. EMBO Rep. 2008, 9, 548–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhäupl, K.; Lindau, C.; Hessel, A.; Wang, Y.; Schütze, C.; Jores, T.; Melchionda, L.; Schönfisch, B.; Kalbacher, H.; Bersch, B.; et al. Structural Basis of Membrane Protein Chaperoning through the Mitochondrial Intermembrane Space. Cell 2018, 175, 1365–1379.e25. [Google Scholar] [CrossRef] [Green Version]
- Valpadashi, A.; Callegari, S.; Linden, A.; Neumann, P.; Ficner, R.; Urlaub, H.; Deckers, M.; Rehling, P. Defining the architecture of the human TIM22 complex by chemical crosslinking. FEBS Lett. 2021, 595, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ou, X.; Wang, X.; Sun, D.; Zhou, X.; Wu, X.; Li, Q.; Li, L. Structure of the mitochondrial TIM22 complex from yeast. Cell Res. 2021, 31, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Wang, Q.; Guan, Z.; Wu, Y.; Shen, C.; Hong, S.; Cao, J.; Zhang, X.; Yan, C.; Yin, P. Cryo-EM structure of the human mitochondrial translocase TIM22 complex. Cell Res. 2021, 31, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Anderson, A.J.; Jackson, T.D.; Palmer, C.S.; De Souza, D.P.; Fujihara, K.M.; Stait, T.; Frazier, A.; Clemons, N.J.; Tull, D.; et al. Function of hTim8a in complex IV assembly in neuronal cells provides insight into pathomechanism underlying Mohr-Tranebjærg syndrome. eLife 2019, 8. [Google Scholar] [CrossRef]
- Rehling, P. Protein Insertion into the Mitochondrial Inner Membrane by a Twin-Pore Translocase. Science 2003, 299, 1747–1751. [Google Scholar] [CrossRef] [Green Version]
- Kovermann, P.; Truscott, K.N.; Guiard, B.; Rehling, P.; Sepuri, N.B.; Müller, H.; E Jensen, R.; Wagner, R.; Pfanner, N. Tim22, the Essential Core of the Mitochondrial Protein Insertion Complex, Forms a Voltage-Activated and Signal-Gated Channel. Mol. Cell 2002, 9, 363–373. [Google Scholar] [CrossRef]
- Callegari, S.; Müller, T.; Schulz, C.; Lenz, C.; Jans, D.C.; Wissel, M.; Opazo, F.; Rizzoli, S.O.; Jakobs, S.; Urlaub, H.; et al. A MICOS–TIM22 Association Promotes Carrier Import into Human Mitochondria. J. Mol. Biol. 2019, 431, 2835–2851. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, L.; Dieterle, M.P.; Doan, K.N.; Mårtensson, C.U.; Floerchinger, A.; Campo, M.L.; Pfanner, N.; Becker, T. Dual Role of Mitochondrial Porin in Metabolite Transport across the Outer Membrane and Protein Transfer to the Inner Membrane. Mol. Cell 2019, 73, 1056–1065.e7. [Google Scholar] [CrossRef] [Green Version]
- Sakaue, H.; Shiota, T.; Ishizaka, N.; Kawano, S.; Tamura, Y.; Tan, K.S.; Imai, K.; Motono, C.; Hirokawa, T.; Taki, K.; et al. Porin Associates with Tom22 to Regulate the Mitochondrial Protein Gate Assembly. Mol. Cell 2019, 73, 1044–1055.e8. [Google Scholar] [CrossRef] [Green Version]
- Mohr, J.; Mageröy, K. Sex-Linked Deafness of a Possibly New Type. Hum. Hered. 1960, 10, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Tranebjaerg, L.; Schwartz, C.; Eriksen, H.; Andreasson, S.; Ponjavic, V.; Dahl, A.; Stevenson, R.E.; May, M.; Arena, F.; Barker, D. A new X linked recessive deafness syndrome with blindness, dystonia, fractures, and mental deficiency is linked to Xq22. J. Med. Genet. 1995, 32, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Paschen, S.A.; Rothbauer, U.; Káldi, K.; Bauer, M.F.; Neupert, W.; Brunner, M. The role of the TIM8-13 complex in the import of Tim23 into mitochondria. EMBO J. 2000, 19, 6392–6400. [Google Scholar] [CrossRef] [Green Version]
- Rothbauer, U.; Hofmann, S.; Mühlenbein, N.; Paschen, S.A.; Gerbitz, K.-D.; Neupert, W.; Brunner, M.; Bauer, M.F. Role of the Deafness Dystonia Peptide 1 (DDP1) in Import of Human Tim23 into the Inner Membrane of Mitochondria. J. Biol. Chem. 2001, 276, 37327–37334. [Google Scholar] [CrossRef] [Green Version]
- Roesch, K.; Curran, S.P.; Tranebjaerg, L.; Koehler, C.M. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 2002, 11, 477–486. [Google Scholar] [CrossRef]
- Sengers, R.; Trijbels, J.; Willems, J.; Daniels, O.; Stadhouders, A. Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J. Pediatr. 1975, 86, 873–880. [Google Scholar] [CrossRef]
- Jordens, E.Z.; Palmieri, L.; Huizing, M.; Heuvel, L.P.V.D.; Sengers, R.C.A.; Dörner, A.; Ruitenbeek, W.; Trijbels, F.J.; Valsson, J.; Sigfusson, G.; et al. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 2002, 52, 95–99. [Google Scholar] [CrossRef]
- Khan, A.O.; Aldahmesh, M.A.; Alkuraya, F.S. Phenotypes of Recessive Pediatric Cataract in a Cohort of Children with Identified Homozygous Gene Mutations (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2015, 113, 7. [Google Scholar]
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, e1800013. [Google Scholar] [CrossRef]
- Paschen, S.A.; Waizenegger, T.; Stan, T.; Preuss, M.; Cyrklaff, M.; Hell, K.; Rapaport, D.; Neupert, W. Evolutionary conservation of biogenesis of β-barrel membrane proteins. Nat. Cell Biol. 2003, 426, 862–866. [Google Scholar] [CrossRef]
- Wiedemann, N.; Kozjak, V.; Chacinska, A.; Schönfisch, B.; Rospert, S.; Ryan, M.T.; Pfanner, N.; Meisinger, C. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nat. Cell Biol. 2003, 424, 565–571. [Google Scholar] [CrossRef]
- Kozjak-Pavlovic, V.; Ross, K.; Benlasfer, N.; Kimmig, S.; Karlas, A.; Rudel, T. Conserved roles of Sam50 and metaxins in VDAC biogenesis. EMBO Rep. 2007, 8, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnert, M.; Wenz, L.-S.; Zerbes, R.M.; Horvath, S.E.; Stroud, D.; Von Der Malsburg, K.; Mueller, J.M.; Oeljeklaus, S.; Perschil, I.; Warscheid, B.; et al. Role of mitochondrial inner membrane organizing system in protein biogenesis of the mitochondrial outer membrane. Mol. Biol. Cell 2012, 23, 3948–3956. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, K.S.; Papić, D.; Schumann, B.; Sperl, D.; Krumpe, K.; Walther, D.M.; Rapaport, D. A crucial role of Mim2 in the biogenesis of mitochondrial outer membrane proteins. J. Cell Sci. 2012, 125, 3464–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papić, D.; Krumpe, K.; Dukanovic, J.; Dimmer, K.S.; Rapaport, D. Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1-dependent import pathway. J. Cell Biol. 2011, 194, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Popov-Čeleketić, J.; Waizenegger, T.; Rapaport, D. Mim1 Functions in an Oligomeric Form to Facilitate the Integration of Tom20 into the Mitochondrial Outer Membrane. J. Mol. Biol. 2008, 376, 671–680. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Keller, M.; Taskin, A.A.; Horvath, S.E.; Guan, X.L.; Prinz, C.; Opalińska, M.; Zorzin, C.; Van Der Laan, M.; Wenk, M.R.; et al. The fusogenic lipid phosphatidic acid promotes the biogenesis of mitochondrial outer membrane protein Ugo1. J. Cell Biol. 2015, 210, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Keskin, A.; Akdoğan, E.; Dunn, C.D. Evidence for Amino Acid Snorkeling from a High-Resolution, In Vivo Analysis of Fis1 Tail-Anchor Insertion at the Mitochondrial Outer Membrane. Genetics 2017, 205, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, T.; Sakaue, H. Multifaceted roles of porin in mitochondrial protein and lipid transport. Biochem. Soc. Trans. 2019, 47, 1269–1277. [Google Scholar] [CrossRef]
- Grevel, A.; Becker, T. Porins as helpers in mitochondrial protein translocation. Biol. Chem. 2019, 401, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Anderson, A.J.; Stojanovski, D. Mitochondrial protein import dysfunction: Mitochondrial disease, neurodegenerative disease and cancer. FEBS Lett. 2021, 595, 1107–1131. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Protein | Import Pathway | Phenotype | Reference |
|---|---|---|---|---|
| p.T265M p.A582V | TOM70 | TOM | anemia, lactic acidosis, and developmental delay | [39] |
| p.T607I | TOM70 | TOM | severe global developmental delay, mild acquired microcephaly, hypotonia, mixed hyperkinetic movement disorder, exaggerated startle response, and irritability | [40] |
| p.I554F | TOM70 | TOM | gross motor impairment, proximal weaknes, spastic ataxia, hypotonia, cogwheeling, truncal titubation, dysmetric motor control, dysarthria, and ptosis | [40] |
| p.R194H | ALR | MIA40 | progressive myopathy and partial combined respiratory-chain deficiency, congenital cataract, sensorineural hearing loss, and developmental delay | [41] |
| p.Q125* p.R194H | ALR | MIA40 | adrenal insufficiency, lactic acidosis, congenital cataracts and respiratory insufficiency secondary to mitochondrial disorder | [42,43] |
| p.C74Afs*76 C259-25_25924delCA p.R194H p.A73Pfs*77 | ALR | MIA40 | progressive muscular hypotonia, congenital cataracts, hypotrophy, and moderate to severe psychomotor delay | [44] |
| p.R201del | AIFM1 | MIA40 | mitochondrial encephalomyopathy | [45] |
| p.G308E | AIFM1 | MIA40 | ventriculomegaly at early gestation | [46] |
| p.E493V | AIFM1 | MIA40 | Cowchock syndrome ( CMTX4) | [47] |
| p.M171I | AIFM1 | MIA40 | Cowchock syndrome ( CMTX4) | [48] |
| p.G262S | AIFM1 | MIA40 | progressive mitochondrial encephalomyopathy | [49] |
| p.V243L | AIFM1 | MIA40 | progressive muscular atrophy, ataxia, hearing loss, and external opthalmoplegia | [50] |
| p.Q479R | AIFM1 | MIA40 | mitochondrial encephalomyopathy, additional complications | [51] |
| p.G338E | AIFM1 | MIA40 | encephalopathy and ventriculomegaly combined with involvement of motor neurons | [52] |
| p.F210L | AIFM1 | MIA40 | isolated late onset axonal polyneuropathy | [53] |
| p.F210S | AIFM1 | MIA40 | early-onset axonal polyneuropathy | [54] |
| p.T260A p.G360R p.R430C p.V498M p.I591M p.A472V p.P475L p.R451Q p.T260A p.L344F p.R422W p.R422Q | AIFM1 | MIA40 | auditory neuropathy spectrum disorder (ANSD) with or without peripheral neuropathy | [55] |
| p.D237G | AIFM1 | MIA40 | spondyloepimetaphyseal dysplasia with mental retardation (SEMD-MR) | [56] |
| p.D237G p.D237V p.Q235H p.D240D c.697-44 T>G (splicing variant) | AIFM1 | MIA40 | hypomyelinating leukodystrophy and spondylometaphyseal dysplasia (H-SMD) | [57] |
| p.M340T p.T141I | AIFM1 | MIA40 | cerebellar ataxia and others | [58] |
| p.E453Q | AIFM1 | MIA40 | ataxic sensory neuropathy and hearing impairment | [59] |
| p.G399S | AIFM1 | MIA40 | cerebellar ataxia and atrophy, mood and behavioural disorder, intellectual disability with or without hearing loss or peripheral neuropathy | [60] |
| p.S349G | AIFM1 | MIA40 | X-linked auditory neuropathy | [61] |
| p.R217W p.T252M | TIM50 | TIM23 | intellectual disability and seizure disorder | [62] |
| p.S112* p.G190A | TIM50 | TIM23 | severe epilepsy and lactic acidosis | [63] |
| p.R114Q p.G269S | TIM50 | TIM23 | visual loss, West syndrome, neutropenia, cardiomyopathy, Leigh syndrome and persistent 3-methylglutaconic aciduria | [64] |
| p.N76D | MAGMAS | TIM23 | early lethal spondylodysplastic dysplasia | [65] |
| p.Q74P | MAGMAS | TIM23 | spondylodysplastic dysplasia | [66] |
| IVS3-1G>C | DNAJC19 | TIM23 | dilated cardiomyopathy with ataxia (DCMA) | [67] |
| p.A100fs*11 | DNAJC19 | TIM23 | dilated cardiomyopathy with ataxia (DCMA) | [68] |
| c.280+1_280+5delGTAAG | DNAJC19 | TIM23 | DCMA combined with progressive cerebellar atrophy | [69] |
| p.Y21* | DNAJC19 | TIM23 | DCMA with sensorineural hearing loss, bilateral basal ganglia lesions | [70] |
| p.D29G | HPS60 | TIM23 | Pelizaeus–Merzbacher disease | [71,72] |
| p.V72I | HPS60 | TIM23 | hereditary spastic paraplegia | [73] |
| p.T320A | HPS60 | TIM23 | familiar dilated cardiomyopathy | [74] |
| p.L73F | HSP10 | TIM23 | neurological and developmental disorder:spasms, hypotonia, developmental delay and macrocephaly | [75] |
| p.A377T p.S96L p.G515R | α-MPP | TIM23 | non-progressive cerebellar ataxia | [76] |
| p.V256M | α-MPP | TIM23 | non-progressive cerebellar ataxia | [77] |
| p.G356S p.A377T | α-MPP | TIM23 | multisystem involvement including profound global developmental delay, severe hypotonia and weakness, respiratory insufficiency, blindness | [78] |
| p.R175C p.A201P p.V177G p.R175H p.I422T | β-MPP | TIM23 | early-onset neurodegenerative disorder:developmental regression, truncal hypotonia, lack of independent ambulation, lack of speech, seizures, ataxia, dystonia | [79] |
| p.L582R p.L71Q p.E602* p.L306F p.K343E p.H512D macro deletion | MIP | TIM23 | left ventricular non-compaction cardiomyopathy, hypotonia, developmental delay | [80] |
| p.R155W | X-Pro aminopeptidase 3 | TIM23 | nephronophthisis associated ciliopathy | [81] |
| 1357G>T c.931_934 delAACA | X-Pro aminopeptidase 3 | TIM23 | kidney disease nephronophthisis | [82] |
| p.Q241Tfs*13 | X-Pro aminopeptidase 3 | TIM23 | early-age nephronophthisis | [83] |
| gene deletion 151delT 183del10 | TIM8A | TIM22 | Mohr-Tranebjaerg syndrome | [84] |
| 108delG | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [85] |
| p.C43Vfs*22 | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [86] |
| p.E24* | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [87] |
| p.R80* | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [88] |
| p.Q38* | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [89] |
| p.Q28* | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [90] |
| p.C66W | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [91] |
| p.M1I | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [92] |
| IVS1-23A>C | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [93] |
| IVS1+1G>A | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [94] |
| IVS1+1G>T | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [95] |
| X-chromosome micro deletions | TIM8A | TIM22 | Mohr–Tranebjaerg syndrome | [96] |
| X-chromosome micro deletions | TIMA8 | TIM22 | Mohr–Tranebjaerg syndrome and X-linked agammaglobulinemia | [97,98,99] |
| c.1131+1G>T p.Y390* c.297 + 2T>C | AGK | TIM22 | myopathy, bilateral cataracts | [100] |
| p.Y102∗ p.R281∗ p.M1I p.Q173∗ p.R138∗ p.G380Lfs∗16 p.M1I p.Y224∗ c.1131 + 5G>A p. Q291Rfs∗8 c.101+?_222-?del c.221+1G>A p.Q405∗ | AGK | TIM22 | Sengers syndrome | [101] |
| p.M1I p.K327* | AGK | TIM22 | Sengers syndrome | [102] |
| p.I175Yfs*2 c.424-1G>A p.R137* p.Q291* p.I346Yfs*39 p.L75Qfs*12 p. R281* c.877+3G>T | AGK | TIM22 | Sengers syndrome | [103] |
| p.I346Yfs*39 | AGK | TIM22 | Sengers syndrome | [104] |
| p.F406Vfs*4 | AGK | TIM22 | Sengers syndrome | [105] |
| Exon 8 splicing variant (p.A142Tfs*4) | AGK | TIM22 | cataracts | [106] |
| p.Y25* p.V33L | TIM22 | TIM22 | hypotonia, gastroesophageal reflux disease, elevated lactate | [107] |
| c.30-1G>A (splicing variant) | MIC13 | TIM22 | severe mitochondrial encephalopathy, recurrent bouts of liver disease | [108] |
| p.G15Efs*75 | MIC13 | TIM22 | mitochondrial encephalopathy | [109] |
| p.W6Pfs*71 | MIC13 | TIM22 | mitochondrial encephalopathy | [110] |
| c.260-2A>G | MIC13 | TIM22 | mitochondrial encephalopathy | [111] |
| p.I117T | MIC26 | TIM22 | progressive developmental delay, lactic acidosis, muscle weakness, hypotonia, weight loss, gastrointestinal and body temperature dysautonomia, repetitive infections, cognitive impairment, autistic behaviour | [112] |
| p.M1L c.544-1G>C splicing variant (p.V182Rfs*3 ) c.208+3_208 + 6del splicing variant(p.A46Vfs*12) p.Y202Ifs*26 p.L99* | MTX2 | SAM | Mandibuloacral dysplasia | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Pesini, E.; Montoya, J.; Pacheu-Grau, D. Molecular Insights into Mitochondrial Protein Translocation and Human Disease. Genes 2021, 12, 1031. https://doi.org/10.3390/genes12071031
Ruiz-Pesini E, Montoya J, Pacheu-Grau D. Molecular Insights into Mitochondrial Protein Translocation and Human Disease. Genes. 2021; 12(7):1031. https://doi.org/10.3390/genes12071031
Chicago/Turabian StyleRuiz-Pesini, Eduardo, Julio Montoya, and David Pacheu-Grau. 2021. "Molecular Insights into Mitochondrial Protein Translocation and Human Disease" Genes 12, no. 7: 1031. https://doi.org/10.3390/genes12071031
APA StyleRuiz-Pesini, E., Montoya, J., & Pacheu-Grau, D. (2021). Molecular Insights into Mitochondrial Protein Translocation and Human Disease. Genes, 12(7), 1031. https://doi.org/10.3390/genes12071031