
No Time to Age: Uncoupling Aging from Chronological Time


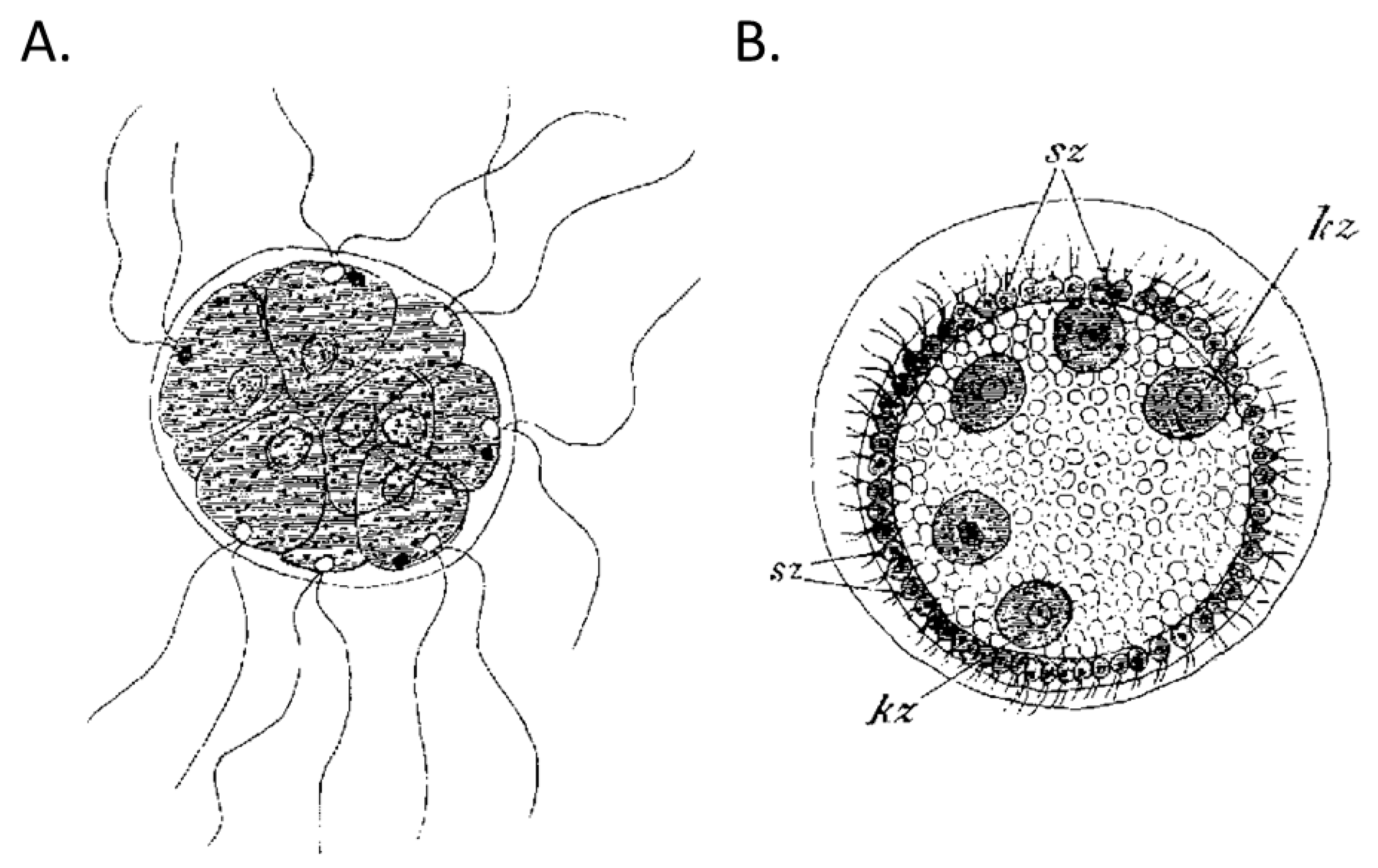{kind=link}
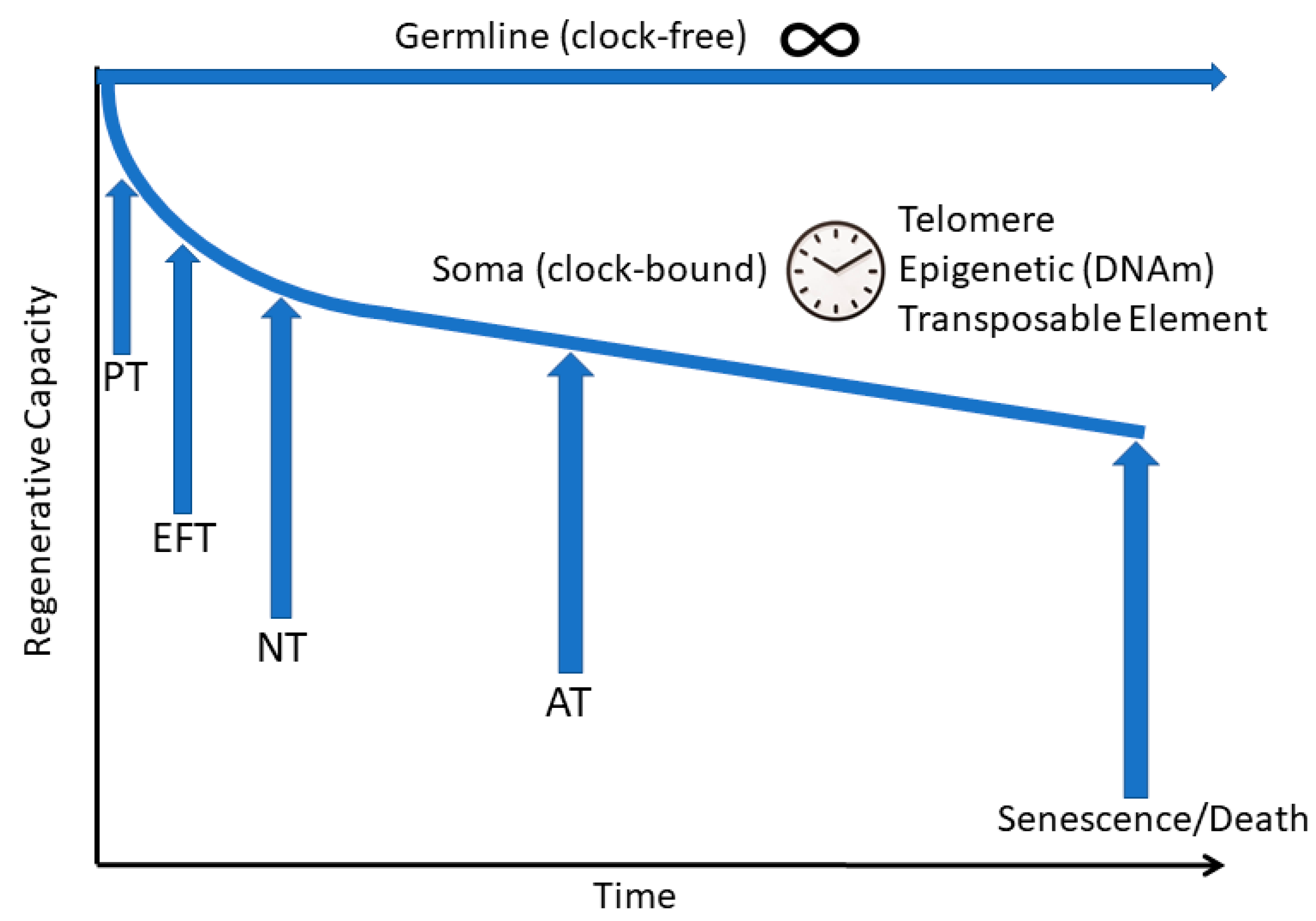{kind=link}
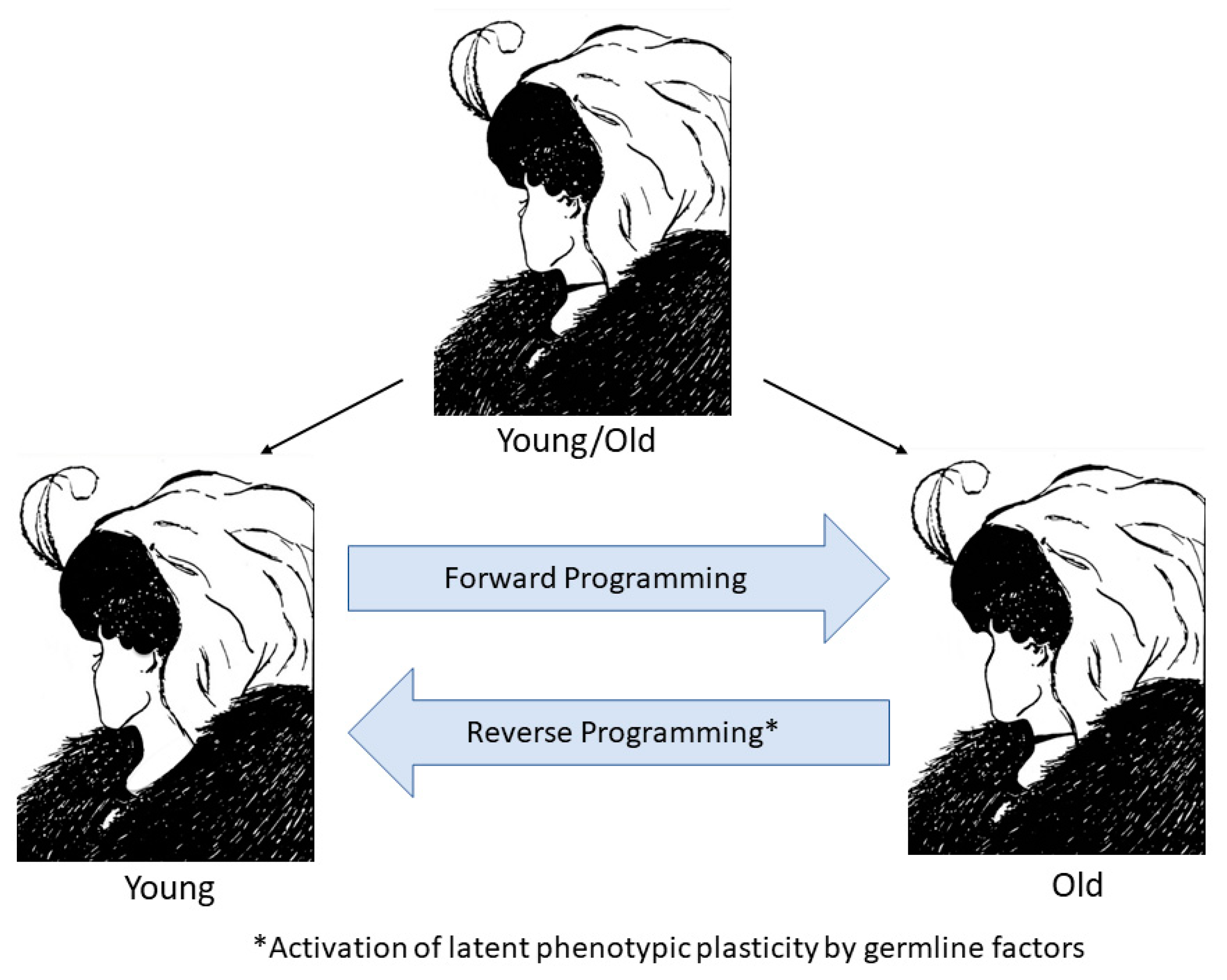{kind=link}
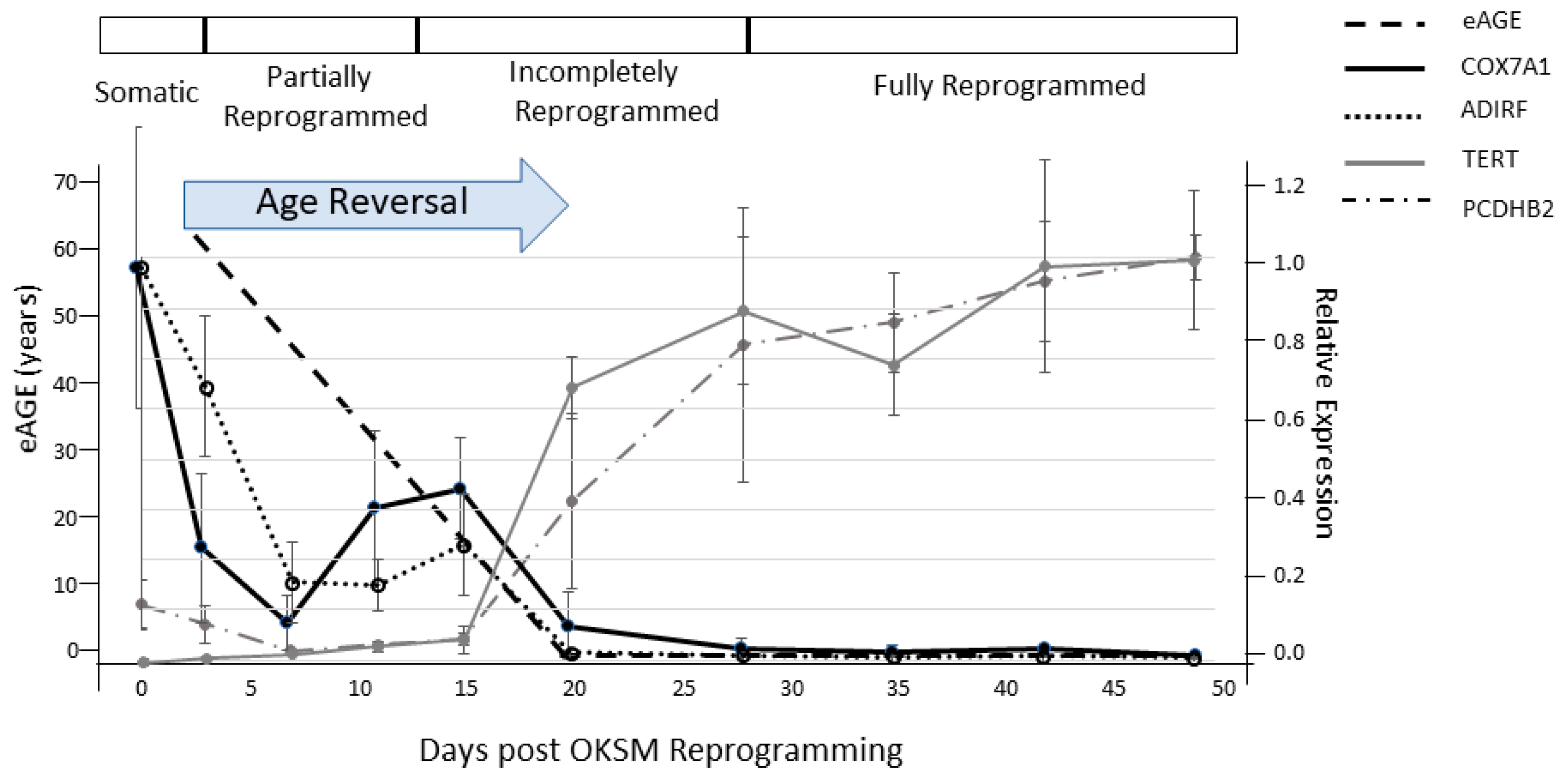{kind=link}
Abstract
:1. Introduction
2. Nature of Somatic Cellular Clocks: Telomeric Clock
3. Nature of Somatic Cellular Clocks: Epigenetic (DNAm) Clock
4. Nature of Somatic Cellular Clocks—Transposable Element Clock
5. Nature of Somatic Cellular Clocks: Aging and Development
6. Uncoupling Biological Time from Chronological Time
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Minot, C.S. The Problem of Age, Growth, and Death; G.P. Putnam Sons: New York, NY, USA, 1907; Volume 21. [Google Scholar]
- Calow, P. Bidder’s hypothesis revisited. Solution to some key problems associated with general molecular theory of ageing. Gerontology 1978, 24, 448–458. [Google Scholar] [CrossRef]
- Williams, G.C. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad.Sci. 2007, 1100, 1–13. [Google Scholar] [CrossRef]
- Finch, C.E. Longevity, Senescence, and the Genome; University of Chicago Press: London, UK, 1994. [Google Scholar]
- Finch, C.E. Update on slow aging and negligible senescence—A mini-review. Gerontology 2009, 55, 307–313. [Google Scholar] [CrossRef]
- West, M.D.; Sternberg, H.; Labat, I.; Janus, J.; Chapman, K.B.; Malik, N.N.; de Grey, A.D.; Larocca, D. Toward a unified theory of aging and regeneration. Regen. Med. 2019, 14, 867–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, A. Essays Upon Heredity and Kindred Biological Problems, 2nd ed.; Clarendon Press: Oxford, UK, 1891. [Google Scholar]
- Weiss, M.C.; Preiner, M.; Xavier, J.C.; Zimorski, V.; Martin, W.F. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLoS Genet. 2018, 14, e1007518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Strehler, B.L.; Mildvan, A.S. General theory of mortality and aging. Science 1960, 132, 14–21. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Witkowski, J.A. Dr. Carrel’s immortal cells. Med. Hist. 1980, 24, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Stanulis-Praeger, B.M. Cellular senescence revisited: A review. Mech. Ageing Dev. 1987, 38, 1–48. [Google Scholar] [CrossRef]
- Dell’Orco, R.T.; Mertens, J.G.; Kruse, P.F., Jr. Doubling potential, calendar time, and senescence of human diploid cells in culture. Exp. Cell Res. 1973, 77, 356–360. [Google Scholar] [CrossRef]
- Olovnikov, A.M. Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk SSSR 1971, 201, 1496–1499. [Google Scholar] [PubMed]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. The immune response and the process of marginotomy in lymphoid cells. Vestn. Akad. Med. Nauk SSSR 1972, 27, 85–87. [Google Scholar]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opresko, P.L.; Shay, J.W. Telomere-associated aging disorders. Ageing Res. Rev. 2017, 33, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B.; Allsopp, R.; Chiu, C.P.; Hausner, M.A.; Hirji, K.; Wang, L.; Harley, C.B.; Villeponteau, B.; West, M.D.; Giorgi, J.V. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS 1996, 10, F17–F22. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Hastie, N.D.; Dempster, M.; Dunlop, M.G.; Thompson, A.M.; Green, D.K.; Allshire, R.C. Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346, 866–868. [Google Scholar] [CrossRef]
- Lindsey, J.; McGill, N.I.; Lindsey, L.A.; Green, D.K.; Cooke, H.J. In vivo loss of telomeric repeats with age in humans. Mutat. Res. 1991, 256, 45–48. [Google Scholar] [CrossRef]
- Harel, I.; Benayoun, B.A.; Machado, B.; Singh, P.P.; Hu, C.K.; Pech, M.F.; Valenzano, D.R.; Zhang, E.; Sharp, S.C.; Artandi, S.E.; et al. A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell 2015, 160, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Shen, J.; Kugan, N.; Furth, E.E.; Lombard, D.B.; Cheung, C.; Pak, S.; Luo, G.; Pignolo, R.J.; DePinho, R.A.; et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol. Cell. Biol. 2004, 24, 8437–8446. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadinanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Parry, E.M.; Li, L.S.; Kembou, F.; Lauder, N.; Hussain, M.A.; Berggren, P.O.; Armanios, M. Short telomeres compromise beta-cell signaling and survival. PLoS ONE 2011, 6, e17858. [Google Scholar]
- Mourkioti, F.; Kustan, J.; Kraft, P.; Day, J.W.; Zhao, M.M.; Kost-Alimova, M.; Protopopov, A.; DePinho, R.A.; Bernstein, D.; Meeker, A.K.; et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol. 2013, 15, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Effros, R.B. From Hayflick to Walford: The role of T cell replicative senescence in human aging. Exp. Gerontol. 2004, 39, 885–890. [Google Scholar] [CrossRef]
- Jacome Burbano, M.S.; Gilson, E. Long-lived post-mitotic cell aging: Is a telomere clock at play? Mech. Ageing Dev. 2020, 189, 111256. [Google Scholar] [CrossRef]
- Phillippe, M.; Phillippe, S.M. Birth and death: Evidence for the same biologic clock. Am. J. Reprod. Immunol. 2017, 77, e12638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittemore, K.; Derevyanko, A.; Martinez, P.; Serrano, R.; Pumarola, M.; Bosch, F.; Blasco, M.A. Telomerase gene therapy ameliorates the effects of neurodegeneration associated to short telomeres in mice. Aging (Albany NY) 2019, 11, 2916–2948. [Google Scholar] [CrossRef]
- Whittemore, K.; Vera, E.; Martinez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef] [Green Version]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. Telomerase reverse transcriptase synergizes with calorie restriction to increase health span and extend mouse longevity. PLoS ONE 2013, 8, e53760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, M.D.; Smith, C.; Kaiser, M.; Levinger, I.; Duque, G.; Gruber, H.J.; Herrmann, M. Physical activity, a modulator of aging through effects on telomere biology. Aging (Albany NY) 2020, 12, 13803–13823. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Wagner, W. Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY) 2011, 3, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [Green Version]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Horvath, S.; Sridhar, A.; Chitsazan, A.; Reh, T.A. Synchrony and asynchrony between an epigenetic clock and developmental timing. Sci. Rep. 2019, 9, 3770. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Choufani, S.; Weksberg, R.; Wilson, S.L.; Yuan, V.; Burt, A.; Marsit, C.; Lu, A.T.; Ritz, B.; Bohlin, J.; et al. Placental epigenetic clocks: Estimating gestational age using placental DNA methylation levels. Aging (Albany NY) 2019, 11, 4238–4253. [Google Scholar] [CrossRef]
- Knight, A.K.; Craig, J.M.; Theda, C.; Baekvad-Hansen, M.; Bybjerg-Grauholm, J.; Hansen, C.S.; Hollegaard, M.V.; Hougaard, D.M.; Mortensen, P.B.; Weinsheimer, S.M.; et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016, 17, 206. [Google Scholar] [CrossRef]
- Bohlin, J.; Haberg, S.E.; Magnus, P.; Reese, S.E.; Gjessing, H.K.; Magnus, M.C.; Parr, C.L.; Page, C.M.; London, S.J.; Nystad, W. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016, 17, 207. [Google Scholar] [CrossRef] [Green Version]
- Simpkin, A.J.; Suderman, M.; Howe, L.D. Epigenetic clocks for gestational age: Statistical and study design considerations. Clin. Epigenet. 2017, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Salas, L.A.; Wiencke, J.K.; Koestler, D.C.; Zhang, Z.; Christensen, B.C.; Kelsey, K.T. Tracing human stem cell lineage during development using DNA methylation. Genome Res. 2018, 28, 1285–1295. [Google Scholar] [CrossRef]
- Soraas, A.; Matsuyama, M.; de Lima, M.; Wald, D.; Buechner, J.; Gedde-Dahl, T.; Soraas, C.L.; Chen, B.; Ferrucci, L.; Dahl, J.A.; et al. Epigenetic age is a cell-intrinsic property in transplanted human hematopoietic cells. Aging Cell 2019, 18, e12897. [Google Scholar] [CrossRef]
- Frobel, J.; Rahmig, S.; Franzen, J.; Waskow, C.; Wagner, W. Epigenetic aging of human hematopoietic cells is not accelerated upon transplantation into mice. bioRxiv 2018, 10, 67. [Google Scholar] [CrossRef]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.K.; Krueger, F.; Team BI, A.C.; von Meyenn, F.; Stegle, O.; Reik, W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY) 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Raj, K.; Horvath, S. Current perspectives on the cellular and molecular features of epigenetic ageing. Exp. Biol. Med. (Maywood) 2020, 245, 1532–1542. [Google Scholar] [CrossRef]
- Snir, S.; Farrell, C.; Pellegrini, M. Human epigenetic ageing is logarithmic with time across the entire lifespan. Epigenetics 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017, 25, 954–960.e956. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.J.; Chwialkowska, K.; Rubbi, L.; Lusis, A.J.; Davis, R.C.; Srivastava, A.; Korstanje, R.; Churchill, G.A.; Horvath, S.; Pellegrini, M. A multi-tissue full lifespan epigenetic clock for mice. Aging (Albany NY) 2018, 10, 2832–2854. [Google Scholar] [CrossRef]
- Wagner, W. Epigenetic aging clocks in mice and men. Genome Biol. 2017, 18, 107. [Google Scholar] [CrossRef]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Zhang, J.; Ablaeva, J.; Adams, D.M.; Almunia, J.; et al. Universal DNA methylation age across mammalian tissues. bioRxiv 2021. [Google Scholar] [CrossRef]
- Vaiserman, A. Developmental epigenetic programming of caste-specific differences in social insects: An impact on longevity. Curr. Aging Sci. 2014, 7, 176–186. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Breitling, L.P.; Saum, K.U.; Perna, L.; Schottker, B.; Holleczek, B.; Brenner, H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenet. 2016, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Kabacik, S.; Horvath, S.; Cohen, H.; Raj, K. Epigenetic ageing is distinct from senescence-mediated ageing and is not prevented by telomerase expression. Aging (Albany NY) 2018, 10, 2800–2815. [Google Scholar] [CrossRef]
- Wagner, W. The Link Between Epigenetic Clocks for Aging and Senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Quach, A.; Moore, D.J.; Achim, C.L.; Soontornniyomkij, V.; Masliah, E.; Singer, E.J.; Gelman, B.; Nemanim, N.; Horvath, S. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J. Neurovirol. 2016, 22, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY) 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Gutman, D.; Rivkin, E.; Fadida, A.; Sharvit, L.; Hermush, V.; Rubin, E.; Kirshner, D.; Sabin, I.; Dwolatzky, T.; Atzmon, G. Exceptionally Long-Lived Individuals (ELLI) Demonstrate Slower Aging Rate Calculated by DNA Methylation Clocks as Possible Modulators for Healthy Longevity. Int. J. Mol. Sci. 2020, 21, 615. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Boeke, J.D. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997, 11, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Oberdoerffer, P.; Sinclair, D.A. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell. Biol. 2007, 8, 692–702. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A.J.; Morello, T.D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014, 5, 5011. [Google Scholar] [CrossRef]
- Ianni, A.; Hoelper, S.; Krueger, M.; Braun, T.; Bober, E. Sirt7 stabilizes rDNA heterochromatin through recruitment of DNMT1 and Sirt1. Biochem. Biophys. Res. Commun. 2017, 492, 434–440. [Google Scholar] [CrossRef]
- Paredes, S.; Angulo-Ibanez, M.; Tasselli, L.; Carlson, S.M.; Zheng, W.; Li, T.M.; Chua, K.F. The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability. J. Biol. Chem. 2018, 293, 11242–11250. [Google Scholar] [CrossRef] [Green Version]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman HE, D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lemos, B. Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 2019, 29, 325–333. [Google Scholar] [CrossRef]
- Sedivy, J.M.; Kreiling, J.A.; Neretti, N.; De Cecco, M.; Criscione, S.W.; Hofmann, J.W.; Zhao, X.; Ito, T.; Peterson, A.L. Death by transposition—The enemy within? Bioessays 2013, 35, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 2013, 5, 867–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.R.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.J. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet. 2006, 2, e205. [Google Scholar] [CrossRef]
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, D.R.; Swigut, T.; Wysocka, J. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. Elife 2018, 7, e35989. [Google Scholar] [CrossRef]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015, 522, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Lenart, P.; Krejci, L. DNA, the central molecule of aging. Mutat. Res. 2016, 786, 1–7. [Google Scholar] [CrossRef]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Orr, W.C. Tightening the connection between transposable element mobilization and aging. Proc. Natl. Acad. Sci. USA 2016, 113, 11069–11070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.G.; Jones, B.C.; Jiang, N.; Chang, C.; Hosier, S.; Wickremesinghe, P.; Garcia, M.; Hartnett, D.A.; Burhenn, L.; Neretti, N.; et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 11277–11282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Sedivy, J.M. Probing the depths of cellular senescence. J. Cell Biol. 2013, 202, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885 e875. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, T.J.; Cavalier, A.N.; Wahl, D. Repetitive elements as a transcriptomic marker of aging: Evidence in multiple datasets and models. Aging Cell 2020, 19, e13167. [Google Scholar] [CrossRef]
- Czech, B.; Malone, C.D.; Zhou, R.; Stark, A.; Schlingeheyde, C.; Dus, M.; Perrimon, N.; Kellis, M.; Wohlschlegel, J.A.; Sachidanandam, R.; et al. An endogenous small interfering RNA pathway in Drosophila. Nature 2008, 453, 798–802. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, Y.; Saito, K.; Kin, T.; Ono, Y.; Asai, K.; Sunohara, T.; Okada, T.N.; Siomi, M.C.; Siomi, H. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 2008, 453, 793–797. [Google Scholar] [CrossRef]
- Lenart, P.; Novak, J.; Bienertova-Vasku, J. PIWI-piRNA pathway: Setting the pace of aging by reducing DNA damage. Mech. Ageing Dev. 2018, 173, 29–38. [Google Scholar] [CrossRef]
- Sturm, A.; Perczel, A.; Ivics, Z.; Vellai, T. The Piwi-piRNA pathway: Road to immortality. Aging Cell 2017, 16, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Senti, K.A.; Jurczak, D.; Sachidanandam, R.; Brennecke, J. piRNA-guided slicing of transposon transcripts enforces their transcriptional silencing via specifying the nuclear piRNA repertoire. Genes Dev. 2015, 29, 1747–1762. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.A.; Yin, H.; Sweeney, S.; Raha, D.; Snyder, M.; Lin, H. A major epigenetic programming mechanism guided by piRNAs. Dev. Cell 2013, 24, 502–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell. Biol. 2011, 12, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Funayama, N.; Nakatsukasa, M.; Mohri, K.; Masuda, Y.; Agata, K. Piwi expression in archeocytes and choanocytes in demosponges: Insights into the stem cell system in demosponges. Evol. Dev. 2010, 12, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Palakodeti, D.; Smielewska, M.; Lu, Y.C.; Yeo, G.W.; Graveley, B.R. The PIWI proteins SMEDWI-2 and SMEDWI-3 are required for stem cell function and piRNA expression in planarians. RNA 2008, 14, 1174–1186. [Google Scholar] [CrossRef] [Green Version]
- Juliano, C.E.; Reich, A.; Liu, N.; Gotzfried, J.; Zhong, M.; Uman, S.; Reenan, R.A.; Wessel, G.M.; Steele, R.E.; Lin, H. PIWI proteins and PIWI-interacting RNAs function in Hydra somatic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Teefy, B.B.; Siebert, S.; Cazet, J.F.; Lin, H.; Juliano, C.E. PIWI-piRNA pathway-mediated transposable element repression in Hydra somatic stem cells. RNA 2020, 26, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Kuo, D.; Nathanson, J.; Satoh, A.; Pao, G.M.; Yeo, G.W.; Bryant, S.V.; Voss, S.R.; Gardiner, D.M.; Hunter, T. Retrotransposon long interspersed nucleotide element-1 (LINE-1) is activated during salamander limb regeneration. Dev. Growth Differ. 2012, 54, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Nelson, M.C.; Brandt, J.E.; Wessman, M.; Mahmud, N.; Weller, K.P.; Hoffman, R. Human CD34(+) stem cells express the hiwi gene, a human homologue of the Drosophila gene piwi. Blood 2001, 97, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, D.; Meusemann, K.; Korb, J. Longevity and transposon defense, the case of termite reproductives. Proc. Natl. Acad. Sci. USA 2018, 115, 5504–5509. [Google Scholar] [CrossRef] [Green Version]
- West, M.D.; Labat, I.; Sternberg, H.; Larocca, D.; Nasonkin, I.; Chapman, K.B.; Singh, R.; Makarev, E.; Aliper, A.; Kazennov, A.; et al. Use of deep neural network ensembles to identify embryonic-fetal transition markers: Repression of COX7A1 in embryonic and cancer cells. Oncotarget 2018, 9, 7796–7811. [Google Scholar] [CrossRef] [Green Version]
- West, M.D.; Labat, I.; Li, J.; Sim, P.; Janus, J.; Mangelson, H.; Sullivan, S.; Liachko, I.; Labhart, P.; Craske, M.; et al. Differential Expression of α, β, and γ Protocadherin Isoforms During Differentiation, Aging, and Cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sidorov, I.; Kimura, M.; Yashin, A.; Aviv, A. Leukocyte telomere dynamics and human hematopoietic stem cell kinetics during somatic growth. Exp. Hematol. 2009, 37, 514–524. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P.; Church, G.M. Genomes optimize reproduction: Aging as a consequence of the developmental program. Physiology (Bethesda) 2005, 20, 252–259. [Google Scholar] [CrossRef]
- Vaiserman, A. Developmental Tuning of Epigenetic Clock. Front. Genet. 2018, 9, 584. [Google Scholar] [CrossRef] [PubMed]
- Laudet, V. The origins and evolution of vertebrate metamorphosis. Curr. Biol. 2011, 21, R726–R737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P.; Holtze, S.; Vyssokikh, M.Y.; Bakeeva, L.E.; Skulachev, M.V.; Markov, A.V.; Hildebrandt, T.B.; Sadovnichii, V.A. Neoteny, Prolongation of Youth: From Naked Mole Rats to “Naked Apes” (Humans). Physiol. Rev. 2017, 97, 699–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyh-Chang, N.; Zhu, H.; Yvanka de Soysa, T.; Shinoda, G.; Seligson, M.T.; Tsanov, K.M.; Nguyen, L.; Asara, J.M.; Cantley, L.C.; Daley, G.Q. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 2013, 155, 778–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: Insights from a successfully aging species. J. Comp. Physiol. B 2008, 178, 439–445. [Google Scholar] [CrossRef]
- Guevara, E.E.; Lawler, R.R.; Staes, N.; White, C.M.; Sherwood, C.C.; Ely, J.J.; Hopkins, W.D.; Bradley, B.J. Age-associated epigenetic change in chimpanzees and humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190616. [Google Scholar] [CrossRef]
- Jylhava, J.; Pedersen, N.L.; Hagg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.H.; Schumacher, B. BiT age: A transcriptome-based aging clock near the theoretical limit of accuracy. Aging Cell 2021, 20, e13320. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L.; Cremer, T. Cytogerontology since 1881: A reappraisal of August Weismann and a review of modern progress. Hum. Genet. 1982, 60, 101–121. [Google Scholar] [CrossRef] [Green Version]
- Gurdon, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar] [PubMed]
- Campbell, K.H.; McWhir, J.; Ritchie, W.A.; Wilmut, I. Sheep cloned by nuclear transfer from a cultured cell line. Nature 1996, 380, 64–66. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Betts, D.; Bordignon, V.; Hill, J.; Winger, Q.; Westhusin, M.; Smith, L.; King, W. Reprogramming of telomerase activity and rebuilding of telomere length in cloned cattle. Proc. Natl. Acad. Sci. USA 2001, 98, 1077–1082. [Google Scholar] [CrossRef]
- Lanza, R.P.; Cibelli, J.B.; Blackwell, C.; Cristofalo, V.J.; Francis, M.K.; Baerlocher, G.M.; Mak, J.; Schertzer, M.; Chavez, E.A.; Sawyer, N.; et al. Extension of cell life-span and telomere length in animals cloned from senescent somatic cells. Science 2000, 288, 665–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, R.P.; Cibelli, J.B.; Faber, D.; Sweeney, R.W.; Henderson, B.; Nevala, W.; West, M.D.; Wettstein, P.J. Cloned cattle can be healthy and normal. Science 2001, 294, 1893–1894. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.Y.; Weick, J.P.; Yu, J.; Ma, L.X.; Zhang, X.Q.; Thomson, J.A.; Zhang, S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Lu, S.J.; Klimanskaya, I.; Gomes, I.; Kim, D.; Chung, Y.; Honig, G.R.; Kim, K.S.; Lanza, R. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells 2010, 28, 704–712. [Google Scholar] [CrossRef]
- Vaziri, H.; Chapman, K.B.; Guigova, A.; Teichroeb, J.; Lacher, M.D.; Sternberg, H.; Singec, I.; Briggs, L.; Wheeler, J.; Sampathkumar, J.; et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen. Med. 2010, 5, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [Green Version]
- Yagi, T.; Kosakai, A.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Nabetani, A.; Ishikawa, F.; Arai, Y.; Hirose, N.; et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS ONE 2012, 7, e41572. [Google Scholar]
- Lee, J.; Bignone, P.A.; Coles, L.S.; Liu, Y.; Snyder, E.; Larocca, D. Induced pluripotency and spontaneous reversal of cellular aging in supercentenarian donor cells. Biochem. Biophys. Res. Commun. 2020, 525, 563–569. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.H.; Liu, X.; Canterel-Thouennon, L.; Li, L.; Edmonson, C.; Rennert, O.M. Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Rep. 2014, 2, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial rejuvenation after induced pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef] [Green Version]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Saric, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Wahlestedt, M.; Erlandsson, E.; Kristiansen, T.; Lu, R.; Brakebusch, C.; Weissman, I.L.; Yuan, J.; Martin-Gonzalez, J.; Bryder, D. Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat. Commun. 2017, 8, 14533. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Peng, H.L.; Zhang, R.; Fu, X.M.; Zhang, G.S. Rejuvenation of Cardiac Tissue Developed from Reprogrammed Aged Somatic Cells. Rejuvenation Res. 2017, 20, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Weissman, A. Die Entstehung der Sexualzellen Bei Den Hydromedusen: Zugleich Ein Betrag zur Kenntniss des Baues und der Lebenserscheinungen Dieser Gruppe; Gustav Fischer Verlag: Jena, Germany, 1883. [Google Scholar]
- Matsumoto, Y.; Piraino, S.; Miglietta, M.P. Transcriptome Characterization of Reverse Development in Turritopsis dohrnii (Hydrozoa, Cnidaria). G3 Genes Genomes Genet. 2019, 9, 4127–4138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, E.V.; Ortega-Recalde, O.; Liu, H.; Lamm, M.S.; Rutherford, K.M.; Cross, H.; Black, M.A.; Kardailsky, O.; Graves, J.A.M.; Hore, T.A.; et al. Stress, novel sex genes, and epigenetic reprogramming orchestrate socially controlled sex change. Sci. Adv. 2019, 5, eaaw7006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlenhaut, N.H.; Jakob, S.; Anlag, K.; Eisenberger, T.; Sekido, R.; Kress, J.; Treier, A.C.; Klugmann, C.; Klasen, C.; Holter, N.I.; et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009, 139, 1130–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, C.K.; Murphy, M.W.; Sarver, A.L.; Griswold, M.D.; Bardwell, V.J.; Zarkower, D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 2011, 476, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2019, 18, e12877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, D.; Parry, A.; Santos, F.; Hernando-Herraez, I.; Stubbs, T.M.; Milagre, I.; Reik, W. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Canamero, M.; Rayon, T.; Ors, I.; Grana, O.; Megias, D.; Dominguez, O.; Martinez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doeser, M.C.; Scholer, H.R.; Wu, G. Reduction of Fibrosis and Scar Formation by Partial Reprogramming In Vivo. Stem Cells 2018, 36, 1216–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Cheng, L. Cocktail of chemical compounds robustly promoting cell reprogramming protects liver against acute injury. Protein Cell 2017, 8, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Agarwal, V.; Agarwal, M.; Singh, M. Exosomes: Structure, Biogenesis, Types and Application in Diagnosis and Gene and Drug Delivery. Curr. Gene Ther. 2020, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko EV, R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Dorronsoro, A.; Santiago, F.E.; Grassi, D.; Zhang, T.; Lai, R.C.; McGowan, S.J.; Angelini, L.; Lavasani, M.; Corbo, L.; Lu, A.; et al. Mesenchymal stem cell-derived extracellular vesicles reduce senescence and extend health span in mouse models of aging. Aging Cell 2021, 20, e13337. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Justice, J.N.; Niedernhofer, L.; Robbins, P.D.; Aroda, V.R.; Espeland, M.A.; Kritchevsky, S.B.; Kuchel, G.A.; Barzilai, N. Development of Clinical Trials to Extend Healthy Lifespan. Cardiovasc. Endocrinol. Metab. 2018, 7, 80–83. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larocca, D.; Lee, J.; West, M.D.; Labat, I.; Sternberg, H. No Time to Age: Uncoupling Aging from Chronological Time. Genes 2021, 12, 611. https://doi.org/10.3390/genes12050611
Larocca D, Lee J, West MD, Labat I, Sternberg H. No Time to Age: Uncoupling Aging from Chronological Time. Genes. 2021; 12(5):611. https://doi.org/10.3390/genes12050611
Chicago/Turabian StyleLarocca, Dana, Jieun Lee, Michael D. West, Ivan Labat, and Hal Sternberg. 2021. "No Time to Age: Uncoupling Aging from Chronological Time" Genes 12, no. 5: 611. https://doi.org/10.3390/genes12050611
APA StyleLarocca, D., Lee, J., West, M. D., Labat, I., & Sternberg, H. (2021). No Time to Age: Uncoupling Aging from Chronological Time. Genes, 12(5), 611. https://doi.org/10.3390/genes12050611