
Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
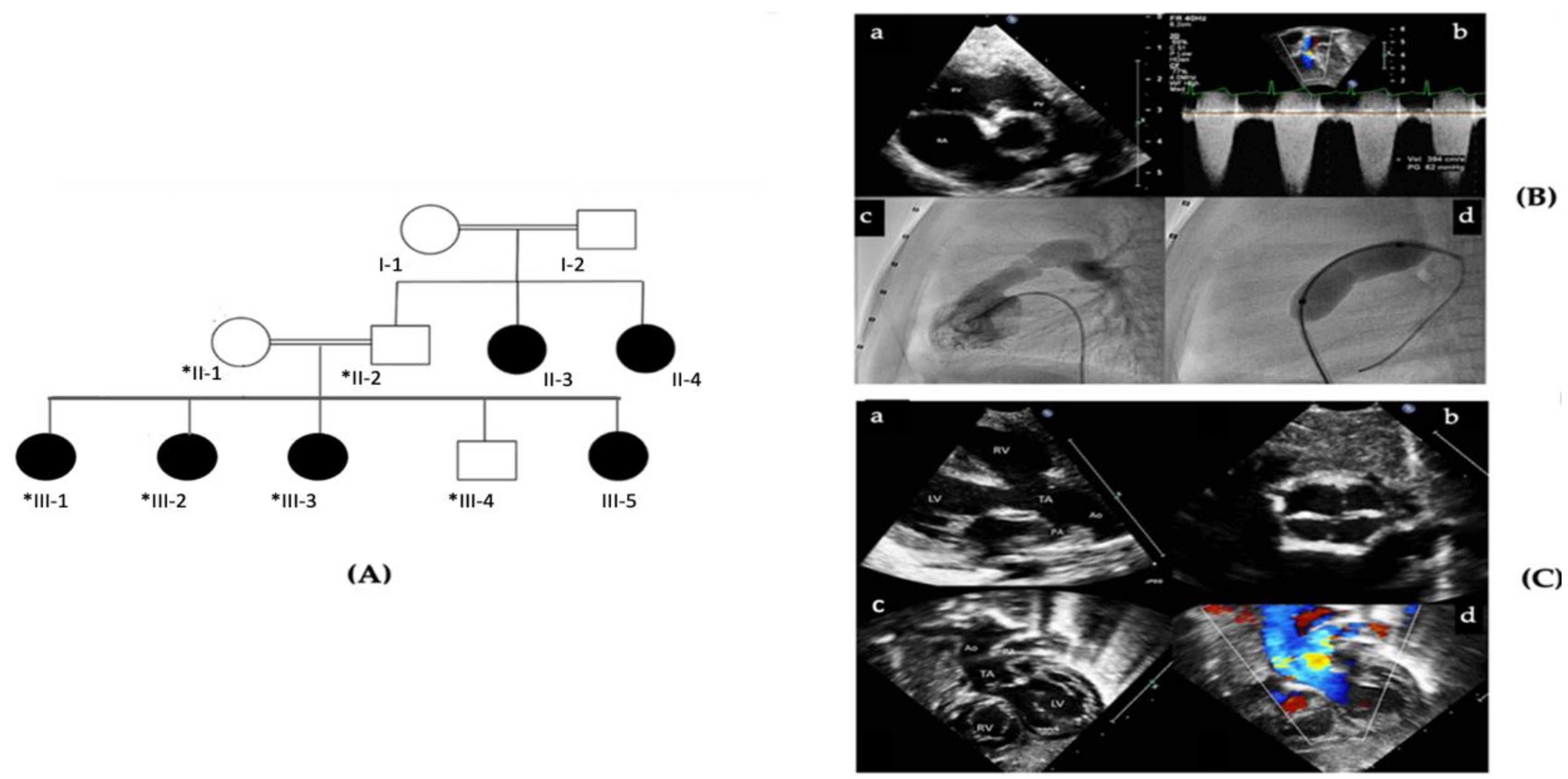
2.1. Patient Description and Ethical Considerations
2.2. Genomic DNA Extraction
2.3. Whole-Exome Sequencing (WES)
2.4. Variant Annotation and Filtering
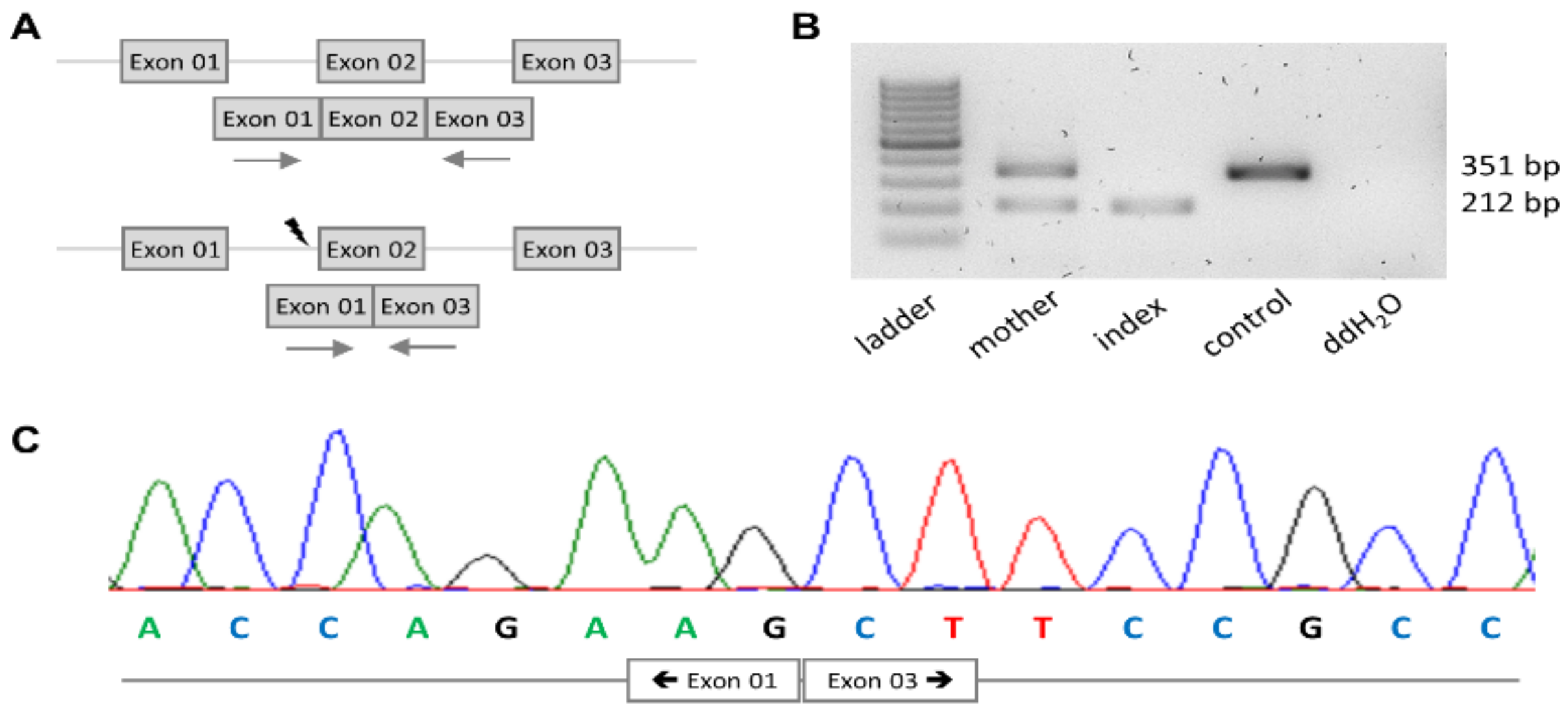
2.5. Sanger Sequencing
2.6. mRNA Analysis
3. Results
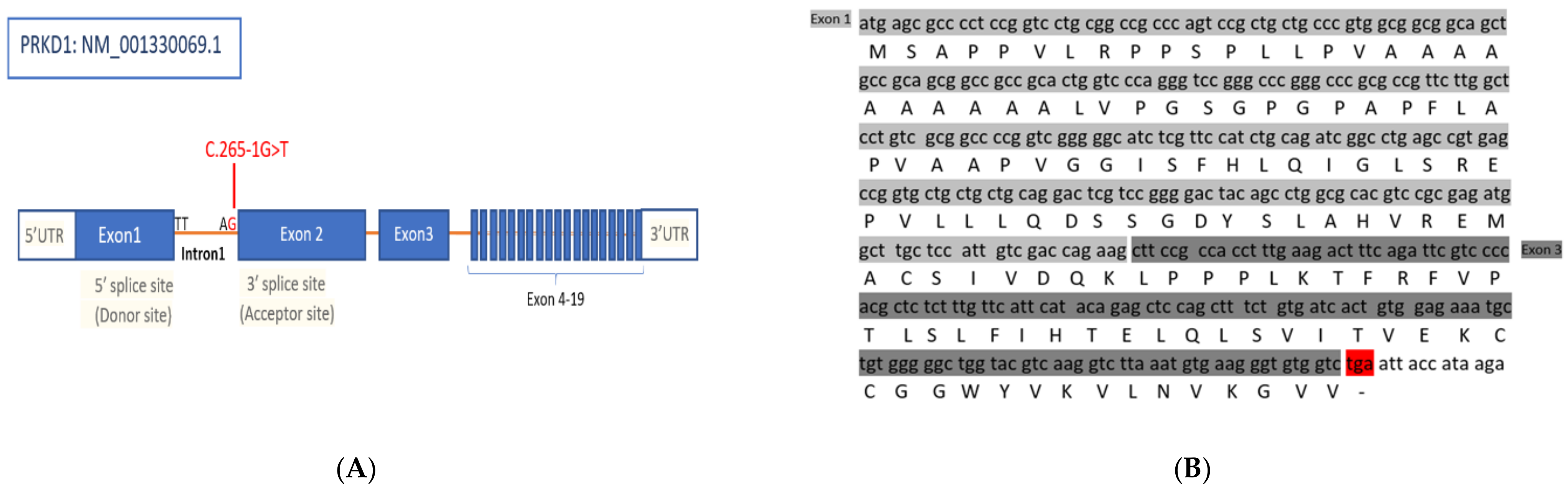
3.1. Identification of a Splice Site Mutation in PRKD1
3.2. PRKD1 Splicing Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LOF | loss-of-function |
| NA | not applicable |
| NR | not reported |
| VSD | Ventricle septal defect |
| ASD | Atrial septal defect |
| AVSD | Atrioventricular septal defect |
| DORV | Double outlet right ventricle |
| LSVC | Left superior vena cava |
| SDS | Sudden death syndrome |
References
- Mendis, S.; Puska, P.; Norrving, B.; Organization, W.H.; Federation, W.H.; Organization, W.S. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011; ISBN 978-92-4-456437-0. [Google Scholar]
- Hoffman, J.I.E.; Kaplan, S. The Incidence of Congenital Heart Disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, S.; Zühlke, L.; Black, G.C.; Choy, M.-K.; Li, N.; Keavney, B.D. Global Birth Prevalence of Congenital Heart Defects 1970–2017: Updated Systematic Review and Meta-Analysis of 260 Studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef]
- Nees, S.N.; Chung, W.K. The Genetics of Isolated Congenital Heart Disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferencz, C.; Rubin, J.D.; McCarter, R.J.; Brenner, J.I.; Neill, C.A.; Perry, L.W.; Hepner, S.I.; Downing, J.W. Congenital Heart Disease: Prevalence at Livebirth. The Baltimore-Washington Infant Study. Am. J. Epidemiol. 1985, 121, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The Developmental Genetics of Congenital Heart Disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef]
- 69 Economic Burden of Congenital Heart Disease on Families|Pediatric Research. Available online: https://www.nature.com/articles/pr2005321 (accessed on 10 March 2021).
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.M.; Al Halees, Z.; Molina, C.; Paterson, R.M. Consanguinity and Congenital Heart Disease in Saudi Arabia. Am. J. Med. Genet. 2001, 99, 8–13. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of Rare Inherited and de Novo Variants in 2871 Congenital Heart Disease Probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Al Hashem, A.; Alghamdi, M.H.; Seidahmad, M.Z.; Wakil, S.M.; Dagriri, K.; Keavney, B.; Goodship, J.; Alyousif, S.; Al-Habshan, F.M.; et al. Positional Mapping of PRKD1, NRP1 and PRDM1 as Novel Candidate Disease Genes in Truncus Arteriosus. J. Med. Genet. 2015, 52, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.T.C.; Bittles, A.H.; Hudgins, L. Consanguinity and the Risk of Congenital Heart Disease. Am. J. Med. Genet. A 2012, 158A, 1236–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massadeh, S.; Alhashem, A.; van de Laar, I.M.B.H.; Alhabshan, F.; Ordonez, N.; Alawbathani, S.; Khan, S.; Kabbani, M.S.; Chaikhouni, F.; Sheereen, A.; et al. ADAMTS19-Associated Heart Valve Defects: Novel Genetic Variants Consolidating a Recognizable Cardiac Phenotype. Clin. Genet. 2020, 98, 56–63. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated MRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2020. [Google Scholar] [CrossRef]
- Hartman, R.J.; Rasmussen, S.A.; Botto, L.D.; Riehle-Colarusso, T.; Martin, C.L.; Cragan, J.D.; Shin, M.; Correa, A. The Contribution of Chromosomal Abnormalities to Congenital Heart Defects: A Population-Based Study. Pediatr. Cardiol. 2011, 32, 1147–1157. [Google Scholar] [CrossRef]
- Soemedi, R.; Wilson, I.J.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of Global Rare Copy-Number Variants to the Risk of Sporadic Congenital Heart Disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Wessels, M.W.; Willems, P.J. Genetic Factors in Non-Syndromic Congenital Heart Malformations. Clin. Genet. 2010, 78, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Nemer, G.M. Genetic Causes of Syndromic and Non-Syndromic Congenital Heart Disease. Mutat. Hum. Genet. Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Butler, T.L.; Esposito, G.; Blue, G.M.; Cole, A.D.; Costa, M.W.; Waddell, L.B.; Walizada, G.; Sholler, G.F.; Kirk, E.P.; Feneley, M.; et al. GATA4 Mutations in 357 Unrelated Patients with Congenital Heart Malformation. Genet. Test Mol. Biomark. 2010, 14, 797–802. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Xie, H.; Zhang, E.; Hong, N.; Fu, Q.; Li, F.; Chen, S.; Yu, Y.; Sun, K. Identification of TBX2 and TBX3 Variants in Patients with Conotruncal Heart Defects by Target Sequencing. Hum. Genom. 2018, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Preuss, C.; Capredon, M.; Wünnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef]
- Armstrong Ehrin, J. Bischoff Joyce Heart Valve Development. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, Y.; Yamagishi, T.; Hokari, S.; Nakamura, H. Mechanisms Involved in Valvuloseptal Endocardial Cushion Formation in Early Cardiogenesis: Roles of Transforming Growth Factor (TGF)-Beta and Bone Morphogenetic Protein (BMP). Anat. Rec. 2000, 258, 119–127. [Google Scholar] [CrossRef]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare Variants in SOX17 Are Associated with Pulmonary Arterial Hypertension with Congenital Heart Disease. Genom. Med. 2018, 10, 56. [Google Scholar] [CrossRef]
- Avkiran, M.; Rowland, A.J.; Cuello, F.; Haworth, R.S. Protein Kinase D in the Cardiovascular System: Emerging Roles in Health and Disease. Circ. Res. 2008, 102, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, W.; Steinberg, S.F. Phos-Tag SDS-PAGE Resolves Agonist- and Isoform-Specific Activation Patterns for PKD2 and PKD3 in Cardiomyocytes and Cardiac Fibroblasts. J. Mol. Cell Cardiol. 2016, 99, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, D.G.; Lyons, G.E.; Martin, J.F.; Olson, E.N. Mef2 Gene Expression Marks the Cardiac and Skeletal Muscle Lineages during Mouse Embryogenesis. Development 1994, 120, 1251–1263. [Google Scholar]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of Protein Kinase D1 for Pathological Cardiac Remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of Mouse Cardiac Morphogenesis and Myogenesis by Transcription Factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Bi, W.; Drake, C.J.; Schwarz, J.J. The Transcription Factor MEF2C-Null Mouse Exhibits Complex Vascular Malformations and Reduced Cardiac Expression of Angiopoietin 1 and VEGF. Dev. Biol. 1999, 211, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.A.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct Genetic Architectures for Syndromic and Nonsyndromic Congenital Heart Defects Identified by Exome Sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Schussler, O.; Gharibeh, L.; Mootoosamy, P.; Murith, N.; Tien, V.; Rougemont, A.-L.; Sologashvili, T.; Suuronen, E.; Lecarpentier, Y.; Ruel, M. Cardiac Neural Crest Cells: Their Rhombomeric Specification, Migration, and Association with Heart and Great Vessel Anomalies. Cell Mol. Neurobiol. 2021, 41, 403–429. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Maddirevula, S.; Kurdi, W.; Alanazy, M.H.; Alkhalidi, H.; Al-Owain, M.; Sulaiman, R.A.; Faqeih, E.; Goljan, E.; Ibrahim, N.; et al. Autozygosity Reveals Recessive Mutations and Novel Mechanisms in Dominant Genes: Implications in Variant Interpretation. Genet. Med. 2017, 19, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsagaris, T.J.; Bustamante, R.A.; Friesendorff, R.A. Familial Heart Disease. Dis. Chest 1967, 52, 153–158. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient II-3, II-4 | Patient III-1 | Patient III-2 | Patient III-3 | |
|---|---|---|---|---|
| Age | NA | 10 | 9 | 6 |
| Gender | Females | Female | Female | Female |
| Weight | NA | 40 | 29.6 | NA |
| Height | NA | 148 | 138.5 | NA |
| Cardiovascular Symptoms | NA | Asymptomatic | Respiratory distress | Asymptomatic |
| Clinical Diagnosis | Septal Defects (not specified) | Pulmonary stenosis | TA type II and VSD | ASD |
| Mental Development | Normal | Normal | Normal | Normal |
| Neurological Abnormalities | Normal | Normal | Normal | Normal |
| Motor development | Normal | Normal | Normal | Normal |
| Primer | Sequence |
|---|---|
| Forward | GCATCTCGTTCCATCTGCAG (Exon01) |
| Reverse | CTCCACAGTGATCACAGAAAGC (Exon03) |
| PRKD1 Variants | Mutation | Inheritance | CHD Classification | Gender of Human Subjects | Clinical Diagnosis | Ref. |
|---|---|---|---|---|---|---|
| NM_001330069.1: c.265-1G>T | Disruption of acceptor splice side of Exon 2 (Exon skipping event lead to premature stop codon)—(LOF) | Recessive | Non-syndromic CHD | Female | PS, Tricuspid regurgitation | This Study |
| Female | TA, VSD | |||||
| Female | ASD | |||||
| NA | Male | Unaffected | ||||
| NA | Male/Female | Healthy control | ||||
| NM_002742.2: c.1852 C>T | Homozygous Truncating Mutations in PRKD1(LOF) | Recessive | Non-syndromic CHD | Two Females | Truncus arteriosus | Shaheen et al., 2015 [11] |
| NM_002742.2: c.1774 G>A | De novo missense mutations p. Gly592Arg (Gain of function mutation) | Dominant | Syndromic CHD | Twins Males | Pulmonary valvar abnormality. Coupled with hypoglycemia, jaundice and hypothermia, Delayed speech and language development, Microcephaly, Bilateral conductive hearing impairment, Ectodermal dysplasia, Lipson syndrome. | Sifrim et al., 2016 [33] |
| AVSD, Hypotonia, Scoliosis, | ||||||
| NM_002742.2: c.896 T>G | De novo missense mutations p. leu299Trp (Gain of function mutation) | Dominant | Syndromic CHD | Male | Attention deficit hyperactivity disorder, Microcephaly, Arnold-Chiari type I, Microcephaly, Nystagmus | |
| Chr14: 30,108,080 G>A | Premature stop codon (p.R243X) | Dominant | NR | Hetrotaxy syndrome, Ebstein anomaly, L-loop corrected transposition | Jin et al., 2017 [10] | |
| Chr14: 30,066,751 G>A | Premature stop codon (p.Q794X) | Dominant | NR | ASD, secundum, patent ductus arteriosus, pulmonary stenosis, valvar | ||
| Chr14: 30,046,444 T>A | Stop-less mutation (p.X913C) | Dominant | NR | Aberrant right subclavian artery, abnormal branching left aortic arch, Aortic stenosis, Bicommissural aortic valve, DORV, LSVC, SDS, tubular hypoplasia of aorta, VSD, | ||
| Chr14: 30,100,011 G>A | Premature Stop codon (p.Q537X) | Dominant | NR | Healthy Control | ||
| ENSG00000184304 G>T | De-novo Missense Mutation | Dominant | ASD, secundum, pulmonary stenosis, Tricuspid stenosis | VSD | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massadeh, S.; Albeladi, M.; Albesher, N.; Alhabshan, F.; Kampe, K.D.; Chaikhouni, F.; Kabbani, M.S.; Beetz, C.; Alaamery, M. Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes 2021, 12, 612. https://doi.org/10.3390/genes12050612
Massadeh S, Albeladi M, Albesher N, Alhabshan F, Kampe KD, Chaikhouni F, Kabbani MS, Beetz C, Alaamery M. Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes. 2021; 12(5):612. https://doi.org/10.3390/genes12050612
Chicago/Turabian StyleMassadeh, Salam, Maha Albeladi, Nour Albesher, Fahad Alhabshan, Kapil Dev Kampe, Farah Chaikhouni, Mohamed S. Kabbani, Christian Beetz, and Manal Alaamery. 2021. "Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease" Genes 12, no. 5: 612. https://doi.org/10.3390/genes12050612
APA StyleMassadeh, S., Albeladi, M., Albesher, N., Alhabshan, F., Kampe, K. D., Chaikhouni, F., Kabbani, M. S., Beetz, C., & Alaamery, M. (2021). Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes, 12(5), 612. https://doi.org/10.3390/genes12050612