
Whole-Exome Sequencing, Proteome Landscape, and Immune Cell Migration Patterns in a Clinical Context of Menkes Disease

 ,
,  , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
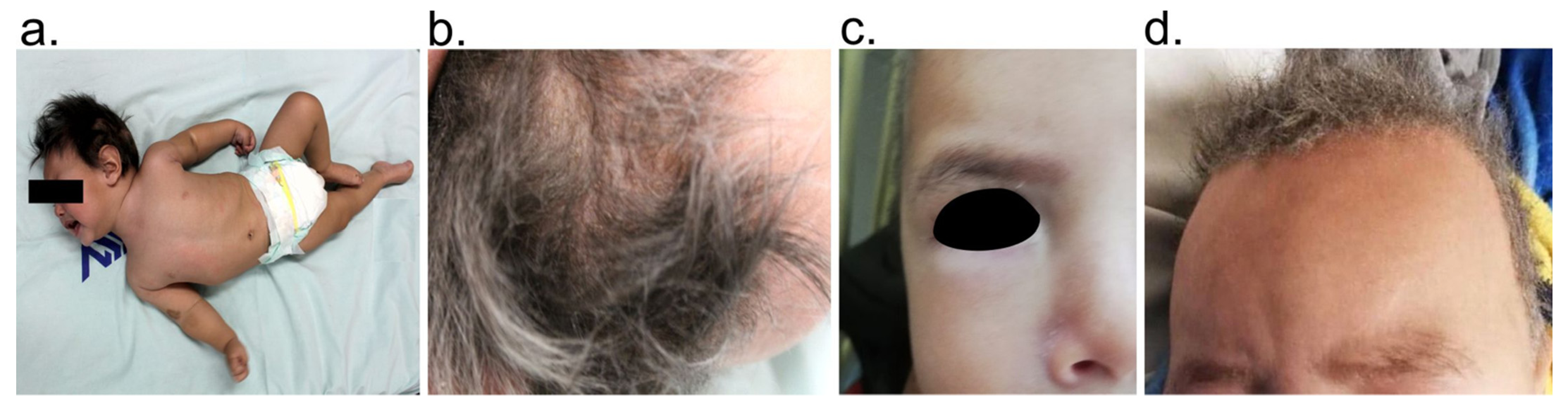
2.2. Clinical Data and Biological Samples
2.3. Nucleic Acid Extraction
2.4. Whole Exome Sequencing
2.5. Plasma Protein Profile Determination by Mass Spectrometry
2.6. Peripheral Blood Cell Migration Assay
2.7. Statistical Analysis
3. Results
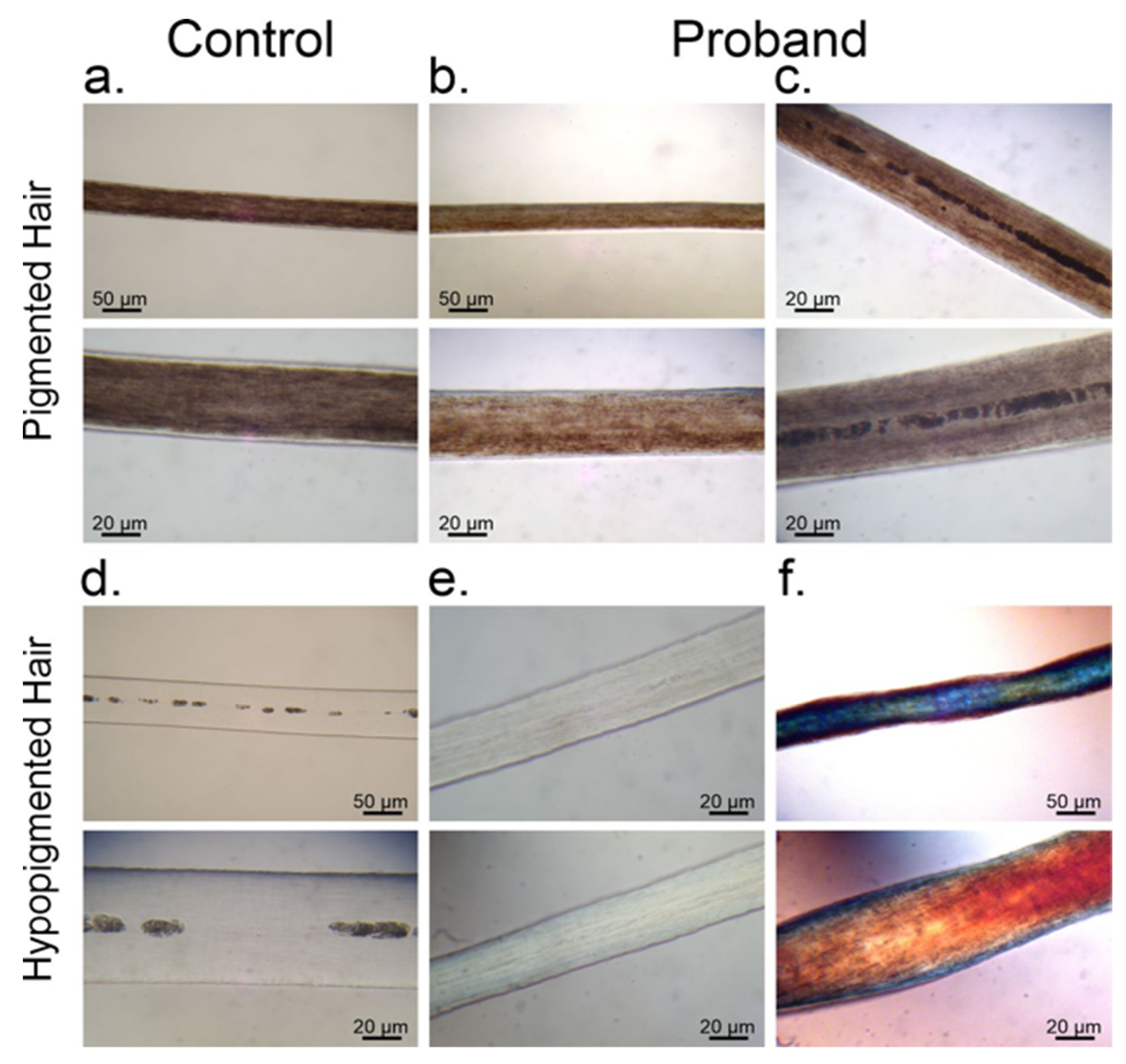
3.1. Melanin Granule Distribution in Scalp Hair Samples
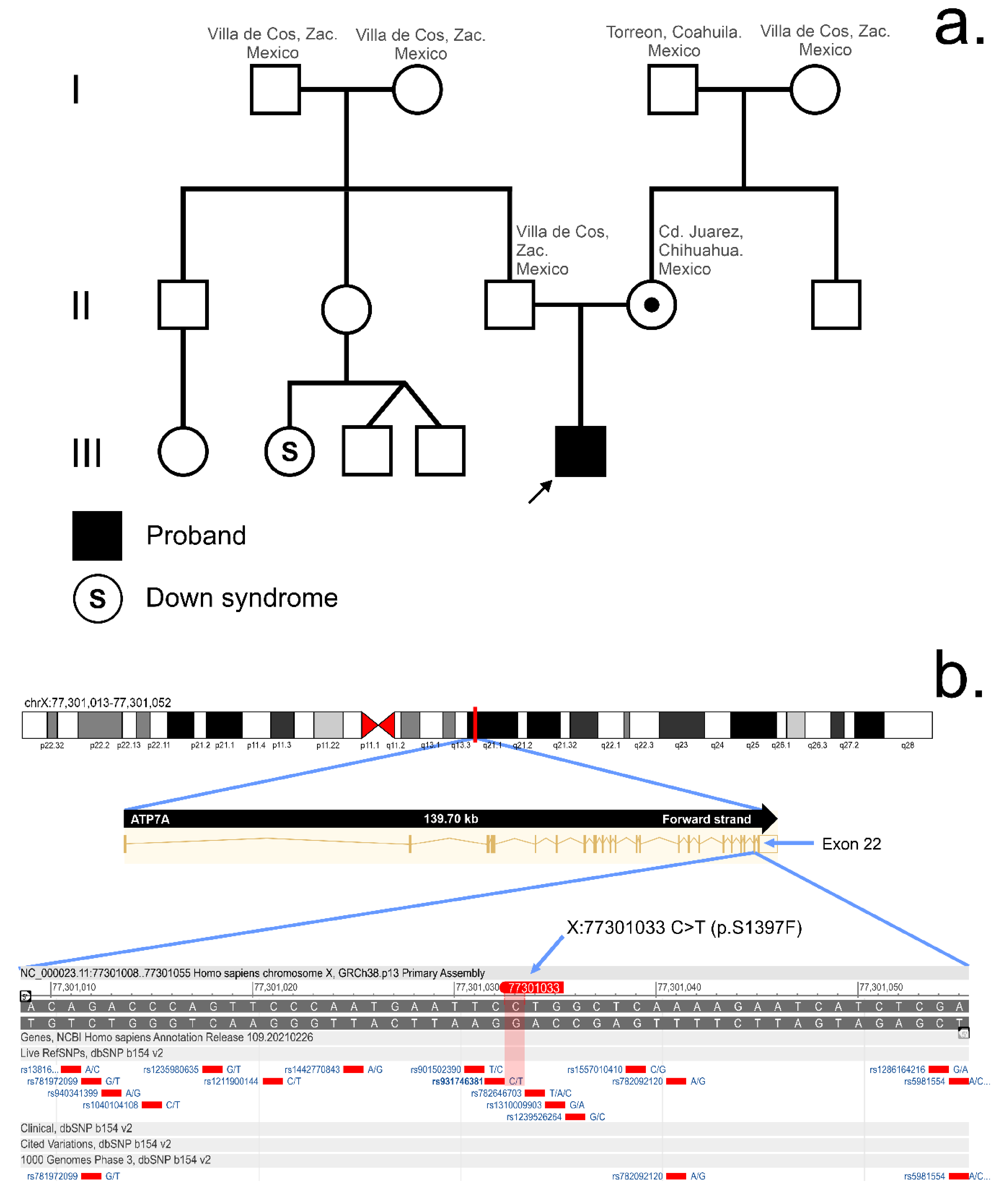
3.2. Whole Exome Sequencing
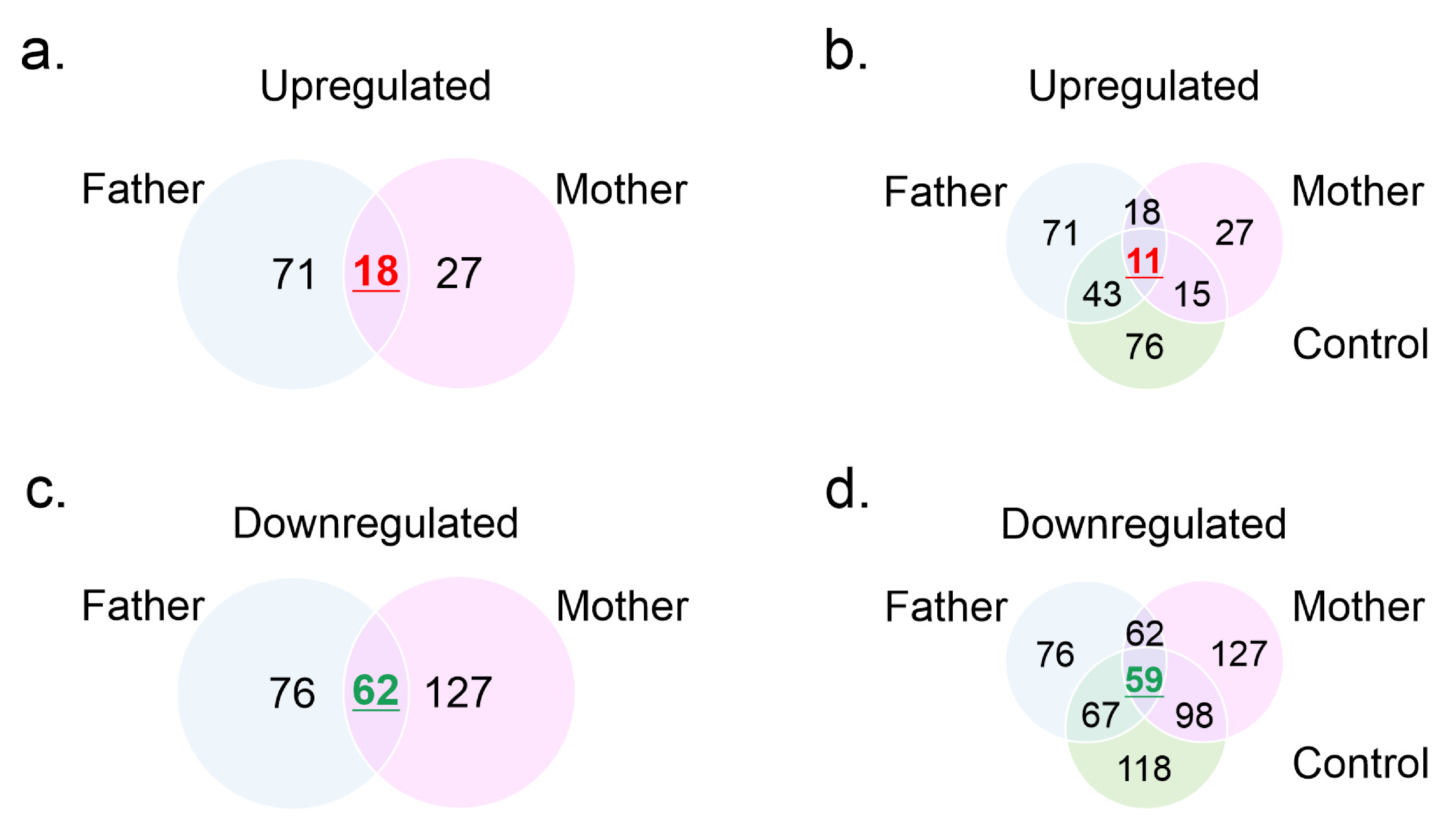
3.3. Mass Spectrometry Profiles
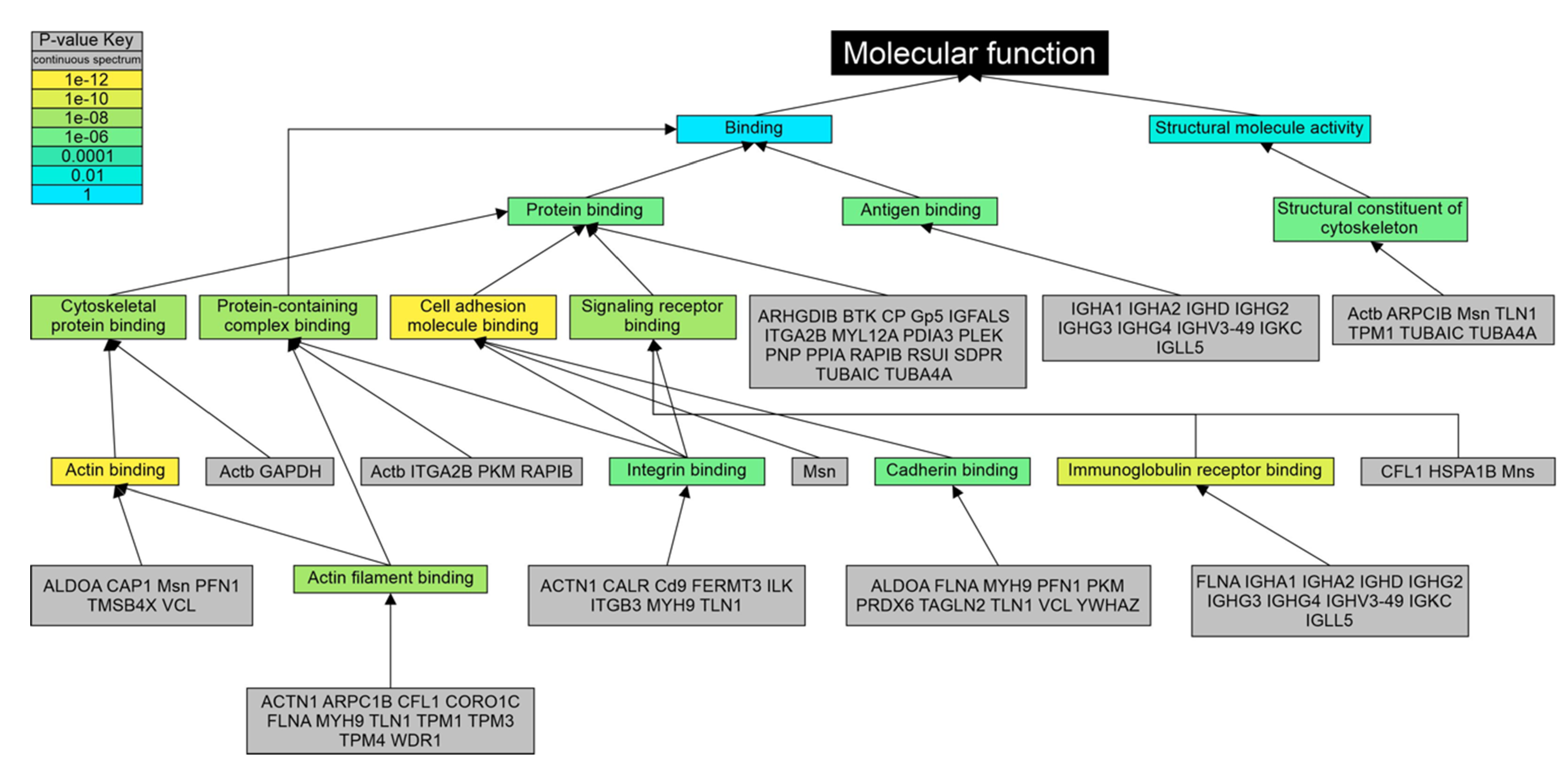
3.4. Gene Ontology Analysis
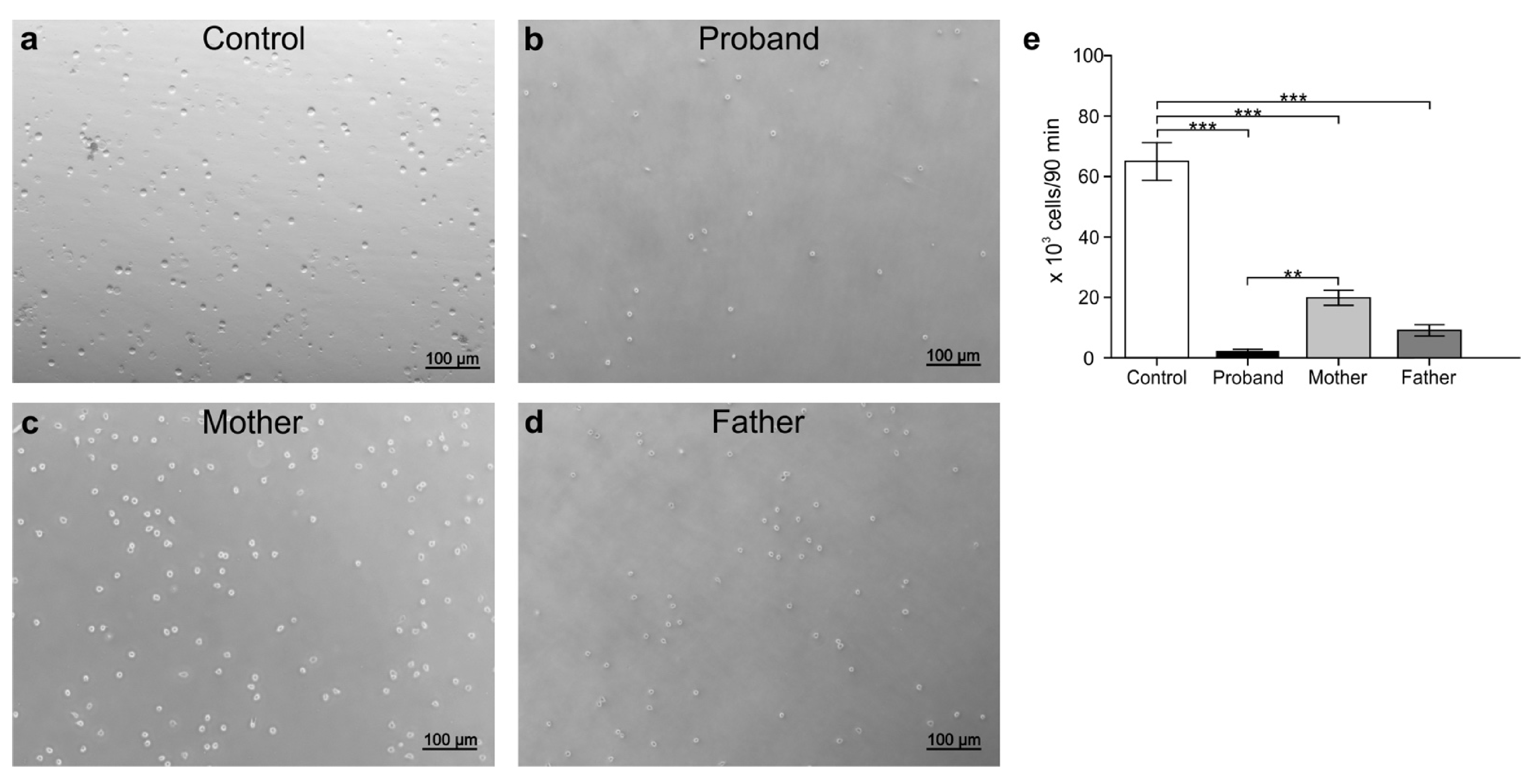
3.5. Peripheral White Blood Cell Migration Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ridaur-Sanz, C.; Durán-McKinster, C.; Rui-Maldonado, R. Usefulness of the skin biopsy as a tool in the diagnosis of silvery hair syndrome. Pediatr. Dermatol. 2018, 35, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Skjørringe, T.; Pedersen, P.A.; Thorborg, S.S.; Nissen, P.; Gourdon, P.; Møller, L.B. Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease. Sci. Rep. 2017, 7, 757. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Prasad, A.N. Menkes disease: What a multidisciplinary approach can do. J. Multidiscip. Healthc. 2016, 9, 371–385. [Google Scholar] [PubMed] [Green Version]
- Møller, L.B.; Bukrinsky, J.T.; Mølgaard, A.; Paulsen, M.; Lund, C.; Tümer, Z.; Larsen, S.; Horn, N. Identification and analysis of 21 novel disease-causing amino acid substitutions in the conserved part of ATP7A. Hum. Mutat. 2005, 26, 84–93. [Google Scholar] [CrossRef]
- Fujisawa, C.; Kodama, H.; Hiroki, T.; Akasaka, Y.; Hamanoue, M. ATP7A mutations in 66 Japanese patients with Menkes disease and carrier detection: A gene analysis. Pediatr. Int. 2019, 61, 345–350. [Google Scholar] [CrossRef]
- de Gemmis, P.; Enzo, M.V.; Lorenzetto, E.; Cattelan, P.; Segat, D.; Hladnik, U. 13 novel putative mutations in ATP7A found in a cohort of 25 Italian families. Metab. Brain. Dis. 2017, 32, 1173–1183. [Google Scholar] [CrossRef]
- Moizard, M.P.; Ronce, N.; Blesson, S.; Bieth, E.; Burglen, L.; Mignot, C.; Mortemousque, I.; Marmin, N.; Dessay, B.; Danesino, C.; et al. Twenty-five novel mutations including duplications in the ATP7A gene. Clin. Genet. 2011, 79, 243–253. [Google Scholar] [CrossRef]
- Cox, D.W. Disorders of copper transport. Br. Med. Bull. 1999, 55, 544–555. [Google Scholar] [CrossRef] [Green Version]
- de Bie, P.; Muller, P.; Wijmenga, C.; Klomp, L.W. Molecular pathogenesis of Wilson and Menkes disease: Correlation of mutations with molecular defects and disease phenotypes. J. Med. Genet. 2007, 44, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Kaler, S.G.; DiStasio, A.T. ATP7A-Related Copper Transport Disorders. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Busti, A. Evidence-Based Medicine Consult. Lab Test: Copper (Blood) Level. Available online: https://www.ebmconsult.com/articles/lab-test-copper-blood-level (accessed on 6 April 2021).
- Physicians Desk Reference. PDR Lab Advisor: A Comprehensive Point-of-Care Guide for Over 600 Lab Tests, 1st ed.; PDR Network, LLC: Montvale, NJ, USA, 2007; pp. 1–500. [Google Scholar]
- Lin, C.N.; Wilson, A.; Church, B.B.; Ehman, S.; Roberts, W.L.; McMillin, G.A. Pediatric reference intervals for serum copper and zinc. Clin. Chim. Acta 2012, 413, 612–615. [Google Scholar] [CrossRef]
- Pagana, K.D.; Pagana, T.J. Mosby’s Manual of Diagnostic and Laboratory Tests, 6th ed.; Elsevier: Philadelphia, PA, USA, 2014; p. 1264. [Google Scholar]
- Busti, A. Evidence-Based Medicine Consult. Lab Test: Ceruloplasmin (Serum) Level. Available online: https://www.ebmconsult.com/articles/lab-test-ceruloplasmin-level (accessed on 6 April 2021).
- Mayo Clinic Laboratories Pediatric Catalog-Ceruloplasmin, Serum. Available online: https://pediatric.testcatalog.org/show/CERS (accessed on 6 April 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; W Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Chin, A.C.; Voss, S.; Parkos, C.A. In vitro neutrophil transepithelial migration. Methods Mol. Biol. 2006, 341, 205–215. [Google Scholar]
- Tümer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mutat. 2013, 34, 417–429. [Google Scholar] [CrossRef]
- Tümer, Z.; Møller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Desai, V.; Donsante, A.; Swoboda, K.J.; Martensen, M.; Thompson, J.; Kaler, S.G. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin. Genet. 2011, 79, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Besold, A.N.; Culbertson, E.M.; Culotta, V.C. The Yin and Yang of copper during infection. J. Biol. Inorg. Chem. 2016, 21, 137–144. [Google Scholar] [CrossRef]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan genomics and the complex architecture of human disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tümer, Z.; Vural, B.; Tønnesen, T.; Chelly, J.; Monaco, A.P.; Horn, N. Characterization of the exon structure of the Menkes disease gene using vectorette PCR. Genomics 1995, 26, 437–442. [Google Scholar] [CrossRef]
- Bonati, M.T.; Verde, F.; Hladnik, U.; Cattelan, P.; Campana, L.; Castronovo, C.; Ticozzi, N.; Maderna, L.; Colombrita, C.; Papa, S. A novel nonsense ATP7A pathogenic variant in a family exhibiting a variable occipital horn syndrome phenotype. Mol. Genet. Metab. Rep. 2017, 13, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Liu, X.Y.; Skjørringe, T.; Morth, J.P.; Møller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Yang, X.; Chen, Y.; Huang, Q.; Wu, Y.; Gu, Q.; Xiao, J.; Yang, H.; Pan, H.; Chen, J.; et al. Identification of novel ATP7A mutations and prenatal diagnosis in Chinese patients with Menkes disease. Metab. Brain. Dis. 2017, 32, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, L.B.; Lenartowicz, M.; Zabot, M.T.; Josiane, A.; Burglen, L.; Bennett, C.; Riconda, D.; Fisher, R.; Janssens, S.; Mohammed, S.; et al. Clinical expression of Menkes disease in females with normal karyotype. Orphanet J. Rare Dis. 2012, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manglani, M.; Adhvaryu, K.; Seth, B. Griscelli syndrome: A case report. Iran J. Child Neurol. 2014, 8, 72–75. [Google Scholar]
- Singh, A.; Garg, A.; Kapoor, S.; Khurana, N.; Entesarian, M.; Tesi, B. An Indian boy with griscelli syndrome type 2: Case report and review of literature. Indian J. Dermatol. 2014, 59, 394–397. [Google Scholar]
- Gonzalez-Mejia, M.E.; Doseff, A.I. Regulation of monocytes and macrophages cell fate. Front. Biosci. 2009, 14, 2413–2431. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Fukata, Y.; Nagata, H.; Miyake, M.; Hama, T. The decreased immune responses in macular mouse, a model of Menkes’ kinky hair disease. Res. Commun. Chem. Pathol. Pharmacol. 1993, 79, 61–73. [Google Scholar]
- Silva, L.C.R.; de Araújo, A.L.; Fernandes, J.R.; Matias, S.T.; Silva, P.R.; Duarte, A.J.S.; Garcez Leme, L.E.; Benard, G. Moderate and intense exercise lifestyles attenuate the effects of aging on telomere length and the survival and composition of T cell subpopulations. Age 2016, 38, 24. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Finding |
|---|---|
| Age (months) | 8 |
| Sex | Male |
| Consanguinity | Absent |
| Ethnicity | Mexican |
| Fever | Absent |
| Abdominal distension | Absent |
| Anasarca | Absent |
| Paleness of body | Present |
| Excessively fair skin | Absent |
| History of similar complaints | Absent |
| Developmental delay | |
| 1. Cephalic support 2. Sedestation | Absent Absent |
| Silvery grey hair | Present |
| Analyte | Reference Values | Proband Age (This Study) | ||
|---|---|---|---|---|
| 0–6 Months | 6–24 Months | 2 Months | 20 Months | |
| Copper [11,12,13,14] | 20–70 µg/dL | 72–178 µg/dL | 60 µg/dL | 90 µg/dL |
| Ceruloplasmin [13,15,16,17] | 5–33 mg/dL | 26–55 mg/dL | 2.5 mg/dL | 16.1 mg/L |
| Finding | Reference Range | 2 Months | 8 Months † | 14 Months | 16 Months | 20 Months |
|---|---|---|---|---|---|---|
| 1. Hematic Biometry | ||||||
| Leucocytes (103/µL) | 6–14 | 10.4 | 6.0 | 19.8 | 15.8 | 11.3 |
| Lymphocytes (%) | 50–60 | 66.8 | 83.1 | 51.7 | 44.5 | 42.7 |
| Monocytes (%) | 2–11 | 0 | 2.7 | 15.0 | 1 | 9 |
| Granulocytes (%) | 33 | 20.4 | 13.4 | 32.2 | 48 | 48.4 |
| Lymphocyte number (103/µL) | 1–9 | 6.9 | 4.96 | 10.2 | 7.1 | 4.8 |
| Monocyte number (103/µL) | <1.0 | 0 | 0.16 | 2.97 | 0.15 | 1.01 |
| Granulocyte number (103/µL) | 1.5–8.5 | 2.2 | 0.80 | 6.39 | 7.74 | 5.5 |
| Red cell blood (106/µL) | 3.8–5.4 | 4.6 | 5.9 | 5.8 | 5.6 | 6.3 |
| Hemoglobin (g/dL) | 10.5–14 | 10.9 | 12.1 | 12.0 | 11.6 | 13.1 |
| Hematocrit (%) | 32–42 | 32.8 | 38.2 | 39.7 | 36.8 | 43.2 |
| Mean corpuscular volume (fl) | 72–88 | 71.5 | 64.3 | 68.3 | 65.2 | 66.8 |
| Mean corpuscular hemoglobin (pg) | 24–30 | 23.7 | 20.4 | 20.7 | 20.6 | 20.7 |
| Red blood cell distribution width (%) | 11.5–16 | 14.3 | 18.6 | 19.2 | 16.1 | 16.7 |
| Platelets (103/µL) | 150–450 | 789 | 220.0 | 177.0 | 435 | 657 |
| Mean platelet volume (Fl) | 6–9.5 | 9.4 | * | * | 10.1 | 9.1 |
| 2. Blood Chemistry Test | ||||||
| Glucose (mg/dL) | 70–110 | 68 | 104 | 72 | 102 | 86 |
| Creatinine (mg/dL) | 0.12–1.06 | 0.33 | 0.36 | 0.37 | 0.21 | 0.39 |
| Urea (mg/dL) | 2.0–6.2 | 20.9 | 17.3 | 39.3 | 1.7 | 22.8 |
| Blood urea nitrogen (mg/dL) | 8–28 | 10 | 8.1 | 18.0 | * | 17 |
| 3. Chemistry Urine Test | ||||||
| pH | 5.5–7.0 | 7.0 | 6.0 | 7.0 | 8.0 | 6.0 |
| Density (mg/dL) | 1.005–1.010 | 1.003 | 1.029 | 1.011 | 1.020 | 1.027 |
| Protein (mg/dL) | negative | negative | 25 | negative | negative | 25 |
| Blood (Ery/mL) | negative | negative | negative | negative | negative | 10 |
| Ketones (mg/dL) | negative | negative | negative | negative | 5 | 15 |
| Glucose (mg/dL) | negative | negative | negative | negative | negative | negative |
| Bilirubin (mg/dL) | negative | negative | negative | negative | negative | negative |
| Urobilinogen (mg/dL) | <2.0 | normal | normal | normal | normal | normal |
| Nitrites (mg/dL) | negative | negative | negative | negative | negative | negative |
| 4. Spleno-hepathomegaly (presence/absence) | – | absence | absence | absence | absence | absence |
| 5. Pancytopenia (yes/not) | – | not | not | not | not | not |
| 6. Bicytopenia (anemia + thrombocytopenia) | – | not | not | not | not | not |
| 7. Intra-citoplasmic granules (leucocytes) | – | not | not | not | not | not |
| 8. Immune dysfunction (B o T cell) | – | yes | yes | yes | yes | yes |
| 9. Hemophagocytosis | – | not | not | not | not | not |
| 10. Triglycerides increased | – | * | * | * | not | * |
| 11. Fibrinogen decreased | – | * | * | not | * | * |
| 12. Decreased albumin | – | not | not | not | not | not |
| 13. Neurological manifestation | ||||||
| Hypotonia | – | yes | yes | yes | yes | yes |
| Seizures | – | yes | not | not | not | not |
| 14. Treatment (supportive) ** | – | yes | yes | yes | yes | yes |
| Gene Name | Protein Name | Ratio | ||
|---|---|---|---|---|
| Proband/Control | Proband/Father | Proband/Mother | ||
| MSN | Moesin | * | * | * |
| IGHG4 | Ig γ-4 chain C region | * | * | * |
| IGHD | Ig delta chain C region | * | * | * |
| IGHA2 | Ig α-2 chain C region | * | * | * |
| PRDX6 | Peroxiredoxin-6 | * | * | * |
| TUBA1C | Tubulin α-1C chain | * | * | * |
| MYL12A | Myosin regulatory light chain 12A | * | * | * |
| MYH9 | Myosin regulatory light chain H9 | * | * | * |
| TUBA4A | Tubulin α-4A chain | * | * | * |
| PNP | Purine nucleoside phosphorylase | * | * | * |
| TPM3 | Tropomyosin α-3 chain | * | * | * |
| RAP1B | Ras-related protein Rap-1b | * | * | * |
| ARPC1B | Actin-related protein 2/3 complex subunit 1B | * | * | * |
| ILK | Integrin-linked protein kinase | * | * | * |
| WDR1 | WD repeat-containing protein 1 | * | * | * |
| SDPR | Serum deprivation-response protein | * | * | * |
| ITGB3 | Integrin β-3 | * | * | * |
| PLEK | Pleckstrin | * | * | * |
| TPM1 | Tropomyosin α-1 chain | * | * | * |
| HSPA1B | Heat shock 70 kDa protein 1B | * | * | * |
| CALR | Calreticulin | * | * | * |
| PDIA3 | Protein disulfide-isomerase A3 | * | * | * |
| BTK | Tyrosine-protein kinase BTK | * | * | * |
| GP5 | Platelet glycoprotein V | * | * | * |
| ARHGDIB | Rho GDP-dissociation inhibitor 2 | * | * | * |
| CAP1 | Adenylyl cyclase-associated protein 1 | * | * | * |
| CORO1C | Coronin-1C | * | * | * |
| ACTN1 | α-actinin-1 | 0.01 | 0.02 | 0.00 |
| IGHG2 | Ig γ-2 chain C region | 0.01 | 0.02 | 0.12 |
| YWHAZ | 14–3-3 protein zeta/delta | 0.02 | 0.02 | 0.005 |
| FLNA | Filamin-A | 0.02 | 0.24 | 0.005 |
| RSU1 | Ras suppressor protein 1 | 0.02 | 0.02 | 0.003 |
| ITGA2B | Integrin α-IIb | 0.03 | 0.03 | 0.004 |
| FERMT3 | Fermitin family homolog 3 | 0.03 | 0.01 | 0.004 |
| IGHG3 | Ig γ-3 chain C region | 0.03 | 0.04 | 0.13 |
| TPM4 | Tropomyosin α-4 chain | 0.03 | 0.03 | 0.01 |
| TLN1 | Talin-1 | 0.04 | 0.24 | 0.01 |
| IGHA1 | Ig α-1 chain C region | 0.04 | 0.09 | 0.28 |
| calm1a | Calmodulin | 0.06 | 0.03 | 0.02 |
| CFL1 | Cofilin-1 | 0.06 | 0.05 | 0.01 |
| VCL | Vinculin | 0.06 | 0.10 | 0.01 |
| IGKC | Ig kappa chain C region | 0.08 | 0.08 | 0.20 |
| TMSB4X | Thymosin β-4 | 0.08 | 0.04 | 0.09 |
| TAGLN2 | Transgelin-2 | 0.09 | 0.05 | 0.01 |
| ACTB | Actin, cytoplasmic 1 | 0.09 | 0.13 | 0.01 |
| IGKV3D-7 | Ig kappa chain V-III region POM | 0.13 | 0.27 | 0.30 |
| IGLL5 | Immunoglobulin lambda-like polypeptide 5 | 0.13 | 0.12 | 0.35 |
| PFN1 | Profilin-1 | 0.14 | 0.12 | 0.02 |
| CP | Ceruloplasmin | 0.15 | 0.11 | 0.15 |
| IGKV3D-11 | Ig kappa chain V-III region VG | 0.16 | 0.41 | 0.46 |
| - | - | 0.16 | 0.13 | 0.35 |
| IGHV3–49 | - | 0.16 | 0.33 | 0.46 |
| CD9 | CD9 antigen | 0.19 | 0.12 | 0.02 |
| F13A1 | Coagulation factor XIII A chain | 0.22 | 0.17 | 0.03 |
| PKM | Pyruvate kinase PKM | 0.23 | 0.30 | 0.03 |
| PPIA | Peptidyl-prolyl cis-trans isomerase A | 0.25 | 0.34 | 0.04 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 0.39 | 0.19 | 0.04 |
| IGFALS | Insulin-like growth factor-binding protein complex acid labile subunit | 0.46 | 0.25 | 0.24 |
| ALDOA | Fructose-bisphosphate aldolase A | 0.48 | 0.42 | 0.04 |
| TNXB | Tenascin-X putative tenascin-XA | 1.73 | ** | ** |
| HGFAC | Hepatocyte growth factor activator | 2.07 | 6.00 | 3.18 |
| PI16 | Peptidase inhibitor 16 | 2.38 | 3.23 | 2.51 |
| HBB | Hemoglobin subunit β | 2.62 | 2.33 | 3.99 |
| MST1 | Hepatocyte growth factor-like protein | 3.02 | 7.70 | 2.63 |
| SHBG | Sex hormone-binding globulin | 3.87 | ** | 2.53 |
| LAMP2 | Lysosome-associated membrane glycoprotein 2 | 4.37 | 2.60 | 5.71 |
| MASP2 | Mannan-binding lectin serine protease 2 | 6.28 | ** | 6.63 |
| PRDX2 | Peroxiredoxin-2 | 7.54 | 7.48 | 2.88 |
| HBG2 | Hemoglobin subunit γ-2 | 12.28 | 5.58 | 15.35 |
| STXBP2 | Syntaxin-binding protein 2 | ** | ** | 163.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Fierro, M.L.; Cabral-Pacheco, G.A.; Garza-Veloz, I.; Acuña-Quiñones, J.; Martinez-de-Villarreal, L.E.; Ibarra-Ramirez, M.; Beuten, J.; Sanchez-Guerrero, S.E.; Villarreal-Martinez, L.; Delgado-Enciso, I.; et al. Whole-Exome Sequencing, Proteome Landscape, and Immune Cell Migration Patterns in a Clinical Context of Menkes Disease. Genes 2021, 12, 744. https://doi.org/10.3390/genes12050744
Martinez-Fierro ML, Cabral-Pacheco GA, Garza-Veloz I, Acuña-Quiñones J, Martinez-de-Villarreal LE, Ibarra-Ramirez M, Beuten J, Sanchez-Guerrero SE, Villarreal-Martinez L, Delgado-Enciso I, et al. Whole-Exome Sequencing, Proteome Landscape, and Immune Cell Migration Patterns in a Clinical Context of Menkes Disease. Genes. 2021; 12(5):744. https://doi.org/10.3390/genes12050744
Chicago/Turabian StyleMartinez-Fierro, Margarita L., Griselda A. Cabral-Pacheco, Idalia Garza-Veloz, Jesus Acuña-Quiñones, Laura E. Martinez-de-Villarreal, Marisol Ibarra-Ramirez, Joke Beuten, Samantha E. Sanchez-Guerrero, Laura Villarreal-Martinez, Ivan Delgado-Enciso, and et al. 2021. "Whole-Exome Sequencing, Proteome Landscape, and Immune Cell Migration Patterns in a Clinical Context of Menkes Disease" Genes 12, no. 5: 744. https://doi.org/10.3390/genes12050744
APA StyleMartinez-Fierro, M. L., Cabral-Pacheco, G. A., Garza-Veloz, I., Acuña-Quiñones, J., Martinez-de-Villarreal, L. E., Ibarra-Ramirez, M., Beuten, J., Sanchez-Guerrero, S. E., Villarreal-Martinez, L., Delgado-Enciso, I., Rodriguez-Sanchez, I. P., Zuñiga-Ramirez, V. Z., Cardenas-Vargas, E., & Romero-Diaz, V. (2021). Whole-Exome Sequencing, Proteome Landscape, and Immune Cell Migration Patterns in a Clinical Context of Menkes Disease. Genes, 12(5), 744. https://doi.org/10.3390/genes12050744