
Genetics and Epigenetics of One-Carbon Metabolism Pathway in Autism Spectrum Disorder: A Sex-Specific Brain Epigenome?

 , , ,
, , ,  , and
, and 
Abstract
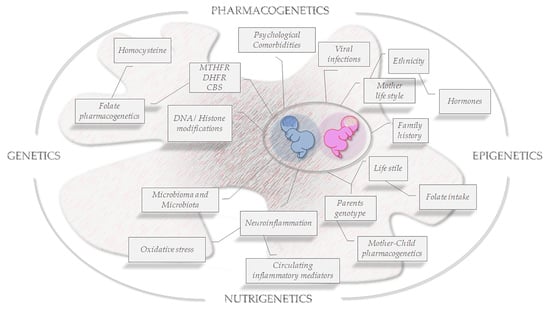
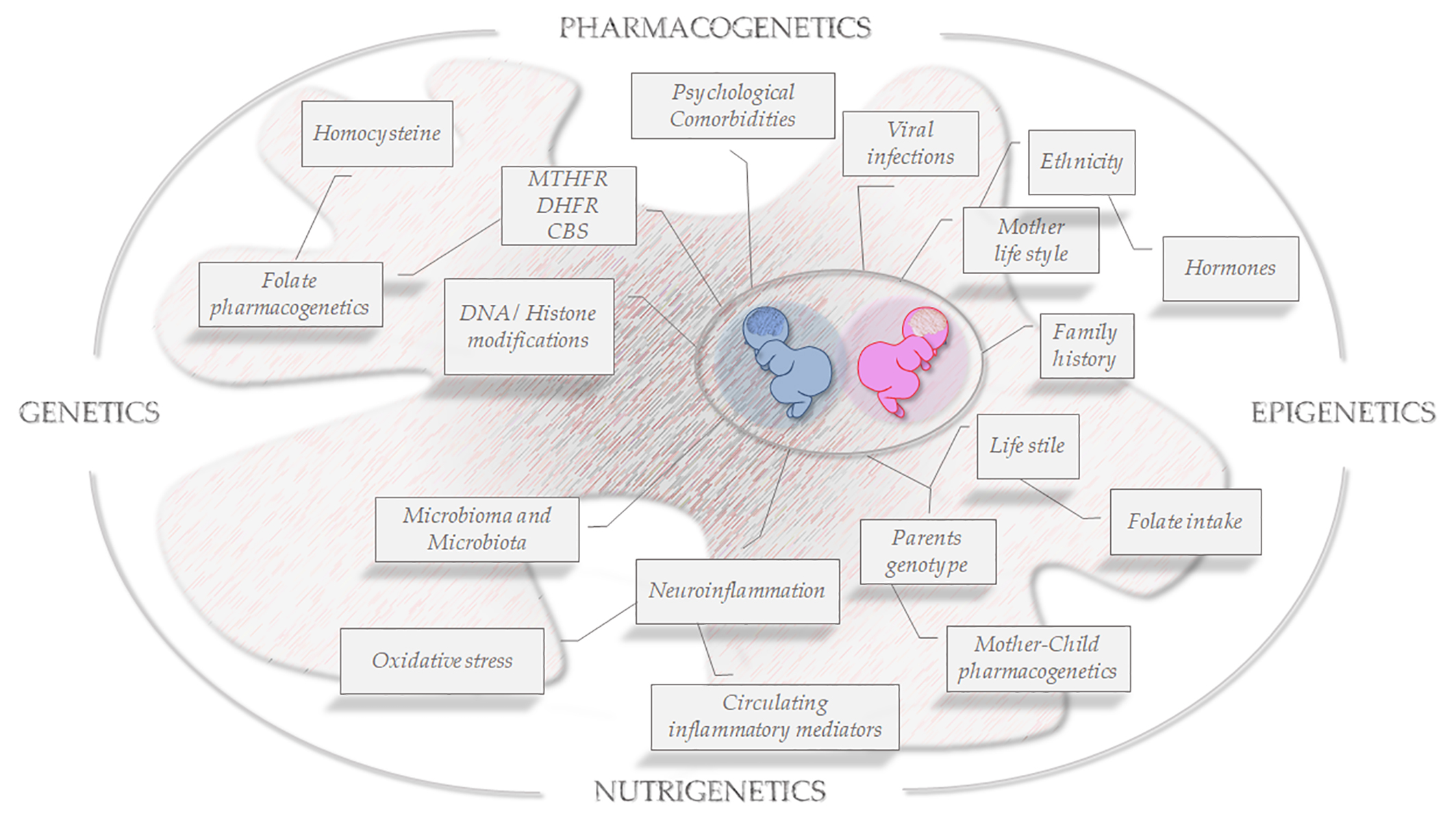
:
1. Introduction
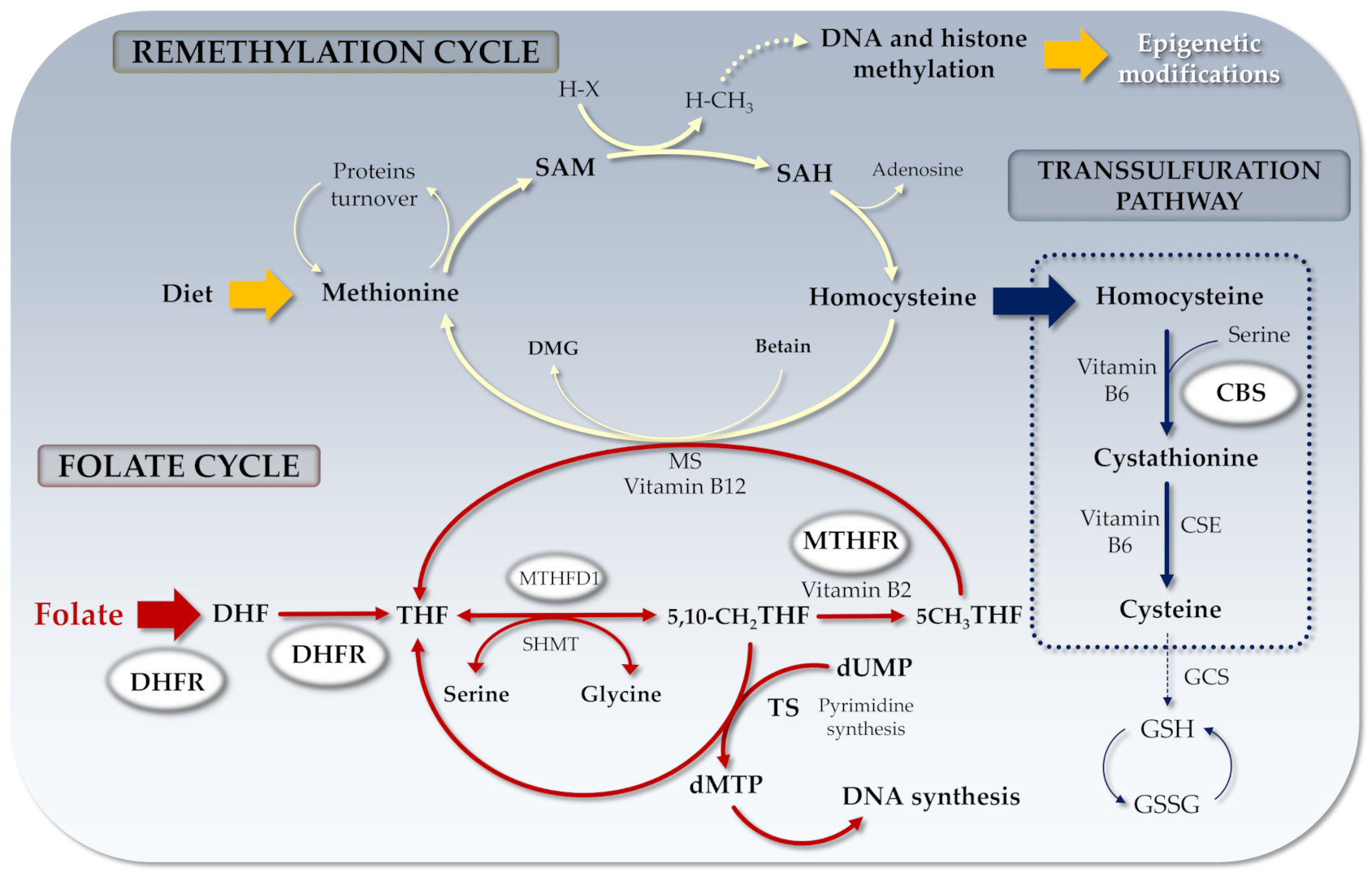
2. One-Carbon Metabolism Pathway
2.1. MTHFR Gene and Functions
2.2. DHFR Gene and Functions
2.3. CBS Gene and Functions
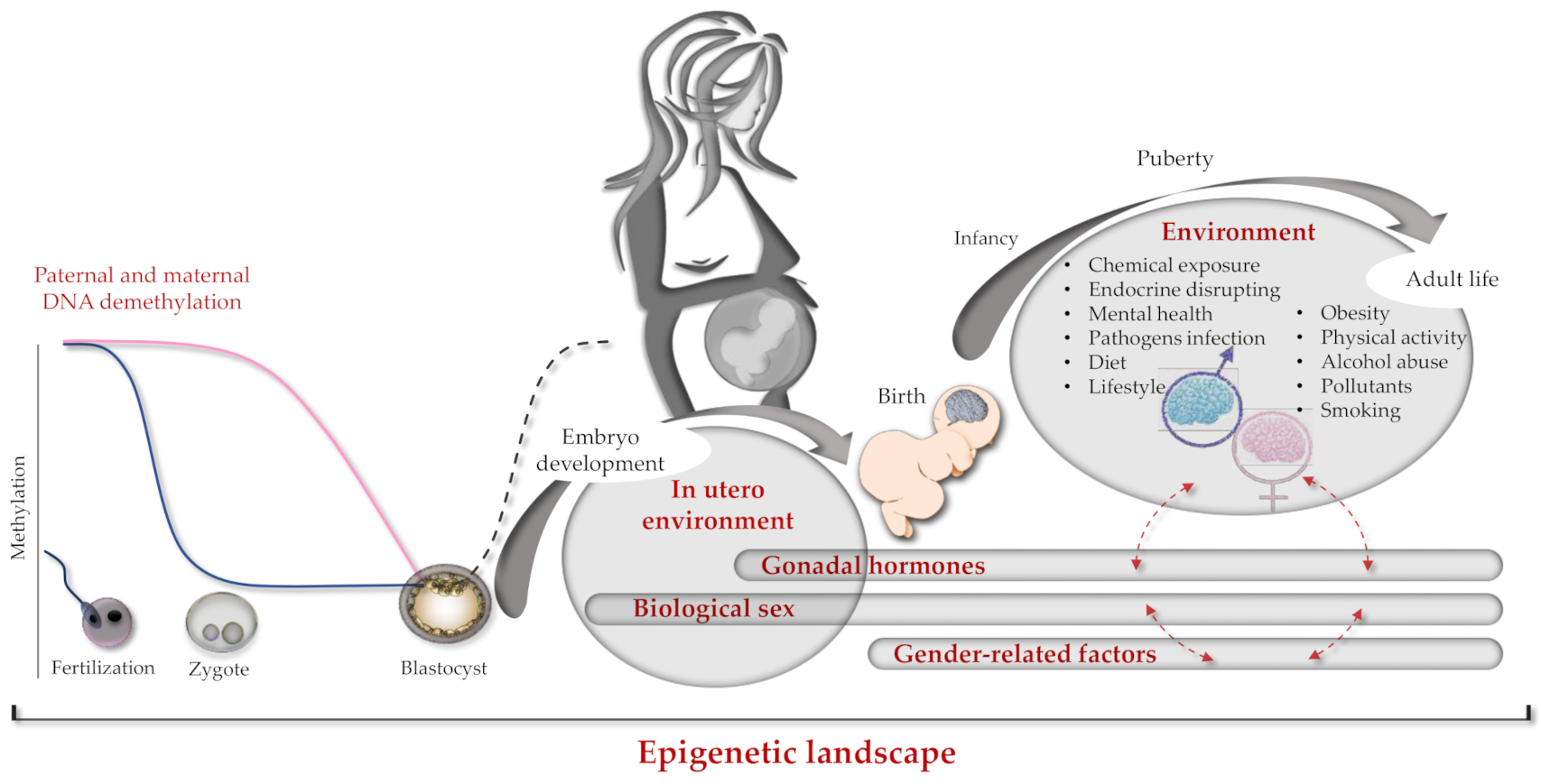
3. One-Carbon Metabolism in Autism and in Other Neurological Diseases: Brain Sex-Related Insights
4. The Role of Epigenetics and Genetics: The Paradigm of the Folate Cycle
5. Conclusions and Future Perspectives in ASD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanner, L. Autistic disturbances of affective contact. Acta Paedopsychiatr. 1968, 35, 100–136. [Google Scholar] [PubMed]
- Bhat, S.; Acharya, U.R.; Adeli, H.; Bairy, G.M.; Adeli, A. Autism: Cause factors, early diagnosis and therapies. Rev. Neurosci. 2014, 25, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Todd, R.D. Autistic traits in the general population: A twin study. Arch. Gen. Psychiatry 2003, 60, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.nimh.nih.gov/health/topics/autism-spectrum-disorders-asd/index.shtml (accessed on 8 March 2021).
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Tisato, V.; Gonelli, A.; Voltan, R.; Secchiero, P.; Zauli, G. Clinical perspectives of TRAIL: Insights into central nervous system disorders. Cell Mol. Life Sci. 2016, 73, 2017–2027. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Verhoeff, T.A.; Perez Pardo, P.; Garssen, J.; Kraneveld, A.D. The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17. Int. J. Mol. Sci. 2020, 22, 118. [Google Scholar] [CrossRef]
- Baranova, J.; Dragunas, G.; Botellho, M.C.S.; Ayub, A.L.P.; Bueno-Alves, R.; Alencar, R.R.; Papaiz, D.D.; Sogayar, M.C.; Ulrich, H.; Correa, R.G. Autism Spectrum Disorder: Signaling Pathways and Prospective Therapeutic Targets. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Melamed, I.R.; Heffron, M.; Testori, A.; Lipe, K. A pilot study of high-dose intravenous immunoglobulin 5% for autism: Impact on autism spectrum and markers of neuroinflammation. Autism Res. 2018, 11, 421–433. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef]
- Guerra, D.J. The molecular genetics of autism spectrum disorders: Genomic mechanisms, neuroimmunopathology, and clinical implications. Autism Res. Treat. 2011, 2011, 398636. [Google Scholar] [CrossRef]
- Razak, K.A.; Dominick, K.C.; Erickson, C.A. Developmental studies in fragile X syndrome. J. Neurodev. Disord. 2020, 12, 13. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Ververi, A.; Dafoulis, V.; Kalyva, E.; Vargiami, E. Autism spectrum disorders: The quest for genetic syndromes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 327–366. [Google Scholar] [CrossRef]
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Nelson, S.; Romano, J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996, 370, 247–261. [Google Scholar] [CrossRef]
- Werling, D.M.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013, 26, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Barrionuevo, F.; Scherer, G. SOX E genes: SOX9 and SOX8 in mammalian testis development. Int. J. Biochem. Cell Biol. 2010, 42, 433–436. [Google Scholar] [CrossRef]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Baekvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Pivina, L.; Semenova, Y.; Dosa, M.D.; Dauletyarova, M.; Bjorklund, G. Iron Deficiency, Cognitive Functions, and Neurobehavioral Disorders in Children. J. Mol. Neurosci. 2019, 68, 1–10. [Google Scholar] [CrossRef]
- Genovese, A.; Butler, M.G. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int. J. Mol. Sci. 2020, 21, 4726. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Zuliani, G.; Vigliano, M.; Longo, G.; Franchini, E.; Secchiero, P.; Zauli, G.; Paraboschi, E.M.; Vikram Singh, A.; Serino, M.L.; et al. Gene-gene interactions among coding genes of iron-homeostasis proteins and APOE-alleles in cognitive impairment diseases. PLoS ONE 2018, 13, e0193867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmati, D.; Varani, K.; Bramanti, B.; Piva, R.; Bonaccorsi, G.; Trentini, A.; Manfrinato, M.C.; Tisato, V.; Care, A.; Bellini, T. “Bridging the Gap” Everything that Could Have Been Avoided If We Had Applied Gender Medicine, Pharmacogenetics and Personalized Medicine in the Gender-Omics and Sex-Omics Era. Int. J. Mol. Sci. 2019, 21, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornese, G.; Iafusco, D.; Monasta, L.; Agnoletto, C.; Tisato, V.; Ventura, A.; Zauli, G.; Secchiero, P. The levels of circulating TRAIL at the onset of type 1 diabetes are markedly decreased in patients with ketoacidosis and with the highest insulin requirement. Acta Diabetol. 2014, 51, 239–246. [Google Scholar] [CrossRef]
- Agostinis, C.; Bulla, R.; Tisato, V.; De Seta, F.; Alberico, S.; Secchiero, P.; Zauli, G. Soluble TRAIL is elevated in recurrent miscarriage and inhibits the in vitro adhesion and migration of HTR8 trophoblastic cells. Hum. Reprod. 2012, 27, 2941–2947. [Google Scholar] [CrossRef] [Green Version]
- Gemmati, D.; Occhionorelli, S.; Tisato, V.; Vigliano, M.; Longo, G.; Gonelli, A.; Sibilla, M.G.; Serino, M.L.; Zamboni, P. Inherited genetic predispositions in F13A1 and F13B genes predict abdominal adhesion formation: Identification of gender prognostic indicators. Sci. Rep. 2018, 8, 16916. [Google Scholar] [CrossRef]
- Myers, S.M.; Voigt, R.G.; Colligan, R.C.; Weaver, A.L.; Storlie, C.B.; Stoeckel, R.E.; Port, J.D.; Katusic, S.K. Autism Spectrum Disorder: Incidence and Time Trends Over Two Decades in a Population-Based Birth Cohort. J. Autism Dev. Disord. 2019, 49, 1455–1474. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Liu, G.; Kay, S.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Sedley, L. Advances in Nutritional Epigenetics-A Fresh Perspective for an Old Idea. Lessons Learned, Limitations, and Future Directions. Epigenet. Insights 2020, 13, 2516865720981924. [Google Scholar] [CrossRef]
- Raghavan, R.; Riley, A.W.; Volk, H.; Caruso, D.; Hironaka, L.; Sices, L.; Hong, X.; Wang, G.; Ji, Y.; Brucato, M.; et al. Maternal Multivitamin Intake, Plasma Folate and Vitamin B12 Levels and Autism Spectrum Disorder Risk in Offspring. Paediatr. Perinat. Epidemiol. 2018, 32, 100–111. [Google Scholar] [CrossRef]
- Naderi, N.; House, J.D. Recent Developments in Folate Nutrition. Adv. Food Nutr. Res. 2018, 83, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.; Fernandez de Cossio, L.; Chakravarty, M.M.; Tremblay, M.E. From Maternal Diet to Neurodevelopmental Disorders: A Story of Neuroinflammation. Front. Cell Neurosci. 2020, 14, 612705. [Google Scholar] [CrossRef]
- Shaik Mohammad, N.; Sai Shruti, P.; Bharathi, V.; Krishna Prasad, C.; Hussain, T.; Alrokayan, S.A.; Naik, U.; Radha Rama Devi, A. Clinical utility of folate pathway genetic polymorphisms in the diagnosis of autism spectrum disorders. Psychiatr. Genet. 2016, 26, 281–286. [Google Scholar] [CrossRef]
- Long, S.; Goldblatt, J. MTHFR genetic testing: Controversy and clinical implications. Aust. Fam. Physician 2016, 45, 237–240. [Google Scholar]
- James, S.J. Autism and Folate-dependent One-carbon Metabolism: Serendipity and Critical Branch-point Decisions in Science. Glob. Adv. Health Med. 2013, 2, 48–51. [Google Scholar] [CrossRef] [Green Version]
- Lintas, C. Linking genetics to epigenetics: The role of folate and folate-related pathways in neurodevelopmental disorders. Clin. Genet. 2019, 95, 241–252. [Google Scholar] [CrossRef]
- Xu, J.; Sinclair, K.D. One-carbon metabolism and epigenetic regulation of embryo development. Reprod. Fertil. Dev. 2015, 27, 667–676. [Google Scholar] [CrossRef]
- Tamura, T.; Picciano, M.F. Folate and human reproduction. Am. J. Clin. Nutr. 2006, 83, 993–1016. [Google Scholar] [CrossRef]
- Desai, A.; Sequeira, J.M.; Quadros, E.V. The metabolic basis for developmental disorders due to defective folate transport. Biochimie 2016, 126, 31–42. [Google Scholar] [CrossRef]
- Lucock, M. Folic acid: Nutritional biochemistry, molecular biology, and role in disease processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef]
- Lan, X.; Field, M.S.; Stover, P.J. Cell cycle regulation of folate-mediated one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1426. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D. Folate-Pathway Gene Variants in Cancer: Haematological Malignancies; Gemmati, D., Ed.; Transworld Research Network: Trivandrum, India, 2008; p. 269. [Google Scholar]
- Field, M.S.; Kamynina, E.; Chon, J.; Stover, P.J. Nuclear Folate Metabolism. Annu. Rev. Nutr. 2018, 38, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Quan, S.; Yang, G.; Ye, Q.; Chen, M.; Yu, H.; Wang, G.; Wang, Y.; Zeng, X.; Qiao, S. One Carbon Metabolism and Mammalian Pregnancy Outcomes. Mol. Nutr. Food Res. 2020, e2000734. [Google Scholar] [CrossRef]
- Gemmati, D.; De Mattei, M.; Catozzi, L.; Della Porta, M.; Serino, M.L.; Ambrosio, C.; Cuneo, A.; Friso, S.; Krampera, M.; Orioli, E.; et al. DHFR 19-bp insertion/deletion polymorphism and MTHFR C677T in adult acute lymphoblastic leukaemia: Is the risk reduction due to intracellular folate unbalancing? Am. J. Hematol. 2009, 84, 526–529. [Google Scholar] [CrossRef]
- Gemmati, D.; Ongaro, A.; Scapoli, G.L.; Della Porta, M.; Tognazzo, S.; Serino, M.L.; Di Bona, E.; Rodeghiero, F.; Gilli, G.; Reverberi, R.; et al. Common gene polymorphisms in the metabolic folate and methylation pathway and the risk of acute lymphoblastic leukemia and non-Hodgkin’s lymphoma in adults. Cancer Epidemiol. Biomark. Prev. 2004, 13, 787–794. [Google Scholar]
- Gemmati, D.; Previati, M.; Serino, M.L.; Moratelli, S.; Guerra, S.; Capitani, S.; Forini, E.; Ballerini, G.; Scapoli, G.L. Low folate levels and thermolabile methylenetetrahydrofolate reductase as primary determinant of mild hyperhomocystinemia in normal and thromboembolic subjects. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Ongaro, A.; De Mattei, M.; Della Porta, M.G.; Rigolin, G.; Ambrosio, C.; Di Raimondo, F.; Pellati, A.; Masieri, F.F.; Caruso, A.; Catozzi, L.; et al. Gene polymorphisms in folate metabolizing enzymes in adult acute lymphoblastic leukemia: Effects on methotrexate-related toxicity and survival. Haematologica 2009, 94, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Tisato, V.; Muggeo, P.; Lupiano, T.; Longo, G.; Serino, M.L.; Grassi, M.; Arcamone, E.; Secchiero, P.; Zauli, G.; Santoro, N.; et al. Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS). Genes 2019, 10, 634. [Google Scholar] [CrossRef] [Green Version]
- Jhee, K.H.; Kruger, W.D. The role of cystathionine β-synthase in homocysteine metabolism. Antioxid. Redox Signal. 2005, 7, 813–822. [Google Scholar] [CrossRef]
- Bhargava, S.; Ali, A.; Bhargava, E.K.; Manocha, A.; Kankra, M.; Das, S.; Mohan Srivastava, L. Lowering homocysteine and modifying nutritional status with folic acid and vitamin B(12) in Indian patients of vascular disease. J. Clin. Biochem. Nutr. 2012, 50, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Margalit, I.; Shochat, T.; Goldberg, E.; Krause, I. Sex Differences in Folate Levels: A Cross Sectional Study of a Large Cohort from Israel. Isr. Med. Assoc. J. 2021, 23, 17–22. [Google Scholar]
- Russo, G.T.; Friso, S.; Jacques, P.F.; Rogers, G.; Cucinotta, D.; Wilson, P.W.; Ordovas, J.M.; Rosenberg, I.H.; Selhub, J.; Framingham Offspring Study, C. Age and gender affect the relation between methylenetetrahydrofolate reductase C677T genotype and fasting plasma homocysteine concentrations in the Framingham Offspring Study Cohort. J. Nutr. 2003, 133, 3416–3421. [Google Scholar] [CrossRef] [Green Version]
- Miles, E.W.; Kraus, J.P. Cystathionine β-synthase: Structure, function, regulation, and location of homocystinuria-causing mutations. J. Biol. Chem. 2004, 279, 29871–29874. [Google Scholar] [CrossRef] [Green Version]
- Gatarek, P.; Rosiak, A.; Borowczyk, K.; Glowacki, R.; Kaluzna-Czaplinska, J. Higher Levels of Low Molecular Weight Sulfur Compounds and Homocysteine Thiolactone in the Urine of Autistic Children. Molecules 2020, 25, 973. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Albero, M.; Cauli, O. Homocysteine Levels in Autism Spectrum Disorder: A Clinical Update. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 289–296. [Google Scholar] [CrossRef]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its Possible Emerging Role in At-Risk Population Groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [Green Version]
- Fulceri, F.; Morelli, M.; Santocchi, E.; Cena, H.; Del Bianco, T.; Narzisi, A.; Calderoni, S.; Muratori, F. Gastrointestinal symptoms and behavioral problems in preschoolers with Autism Spectrum Disorder. Dig. Liver Dis. 2016, 48, 248–254. [Google Scholar] [CrossRef]
- Sun, C.; Zou, M.; Zhao, D.; Xia, W.; Wu, L. Efficacy of Folic Acid Supplementation in Autistic Children Participating in Structured Teaching: An Open-Label Trial. Nutrients 2016, 8, 337. [Google Scholar] [CrossRef] [Green Version]
- Jozefczuk, J.; Kasprzycka, W.; Czarnecki, R.; Graczyk, A.; Jozefczuk, P.; Magda, K.; Lampart, U. Homocysteine as a Diagnostic and Etiopathogenic Factor in Children with Autism Spectrum Disorder. J. Med. Food 2017, 20, 744–749. [Google Scholar] [CrossRef]
- Frye, R.E.; Slattery, J.C.; Quadros, E.V. Folate metabolism abnormalities in autism: Potential biomarkers. Biomark. Med. 2017, 11, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaevitz, L.R.; Berger-Sweeney, J.E. Gene-environment interactions and epigenetic pathways in autism: The importance of one-carbon metabolism. ILAR J. 2012, 53, 322–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, S.C.; Gupta, E.D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Zhu, C.; Zheng, K.; Tang, W.; Gao, L.L.; Trihn, T.H.; Emily Wu, H.; Chen, D.C.; Hong Xiu, M.; Yang Zhang, X. MTHFR Ala222Val polymorphism and clinical characteristics confer susceptibility to suicide attempt in chronic patients with schizophrenia. Sci. Rep. 2020, 10, 5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Yang, B.; Zhi, X.; Wang, Y.; Zheng, Q.; Sun, G. Combined genotype and haplotype distributions of MTHFR C677T and A1298C polymorphisms: A cross-sectional descriptive study of 13,473 Chinese adult women. Medicine (Baltim.) 2016, 95, e5355. [Google Scholar] [CrossRef]
- DeVilbiss, E.A.; Gardner, R.M.; Newschaffer, C.J.; Lee, B.K. Maternal folate status as a risk factor for autism spectrum disorders: A review of existing evidence. Br. J. Nutr. 2015, 114, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Pu, D.; Shen, Y.; Wu, J. Association between MTHFR gene polymorphisms and the risk of autism spectrum disorders: A meta-analysis. Autism Res. 2013, 6, 384–392. [Google Scholar] [CrossRef]
- Mohammad, N.S.; Jain, J.M.; Chintakindi, K.P.; Singh, R.P.; Naik, U.; Akella, R.R. Aberrations in folate metabolic pathway and altered susceptibility to autism. Psychiatr. Genet. 2009, 19, 171–176. [Google Scholar] [CrossRef]
- Cunha, A.L.; Hirata, M.H.; Kim, C.A.; Guerra-Shinohara, E.M.; Nonoyama, K.; Hirata, R.D. Metabolic effects of C677T and A1298C mutations at the MTHFR gene in Brazilian children with neural tube defects. Clin. Chim. Acta 2002, 318, 139–143. [Google Scholar] [CrossRef]
- Xin, Y.; Wu, L.; Lu, X.; Shangguan, S.; Wang, Z.; Chang, S.; Yin, J.; Piao, W.; Zhang, T.; Wang, L. Effects of MTHFR A1298C polymorphism on peripheral blood folate concentration in healthy populations: A meta-analysis of observational studies. Asia Pac. J. Clin. Nutr. 2018, 27, 718–727. [Google Scholar] [CrossRef]
- Stevens, A.J.; Rucklidge, J.J.; Kennedy, M.A. Epigenetics, nutrition and mental health. Is there a relationship? Nutr. Neurosci. 2018, 21, 602–613. [Google Scholar] [CrossRef]
- Isotalo, P.A.; Donnelly, J.G. Prevalence of methylenetetrahydrofolate reductase mutations in patients with venous thrombosis. Mol. Diagn. 2000, 5, 59–66. [Google Scholar] [CrossRef]
- Ogino, S.; Wilson, R.B. Genotype and haplotype distributions of MTHFR677C>T and 1298A>C single nucleotide polymorphisms: A meta-analysis. J. Hum. Genet. 2003, 48, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.J.; Zammit, S.; Gunnell, D.; Smith, G.D. A meta-analysis of the MTHFR C677T polymorphism and schizophrenia risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 135B, 2–4. [Google Scholar] [CrossRef]
- Wan, L.; Li, Y.; Zhang, Z.; Sun, Z.; He, Y.; Li, R. Methylenetetrahydrofolate reductase and psychiatric diseases. Transl. Psychiatry 2018, 8, 242. [Google Scholar] [CrossRef]
- Orenbuch, A.; Fortis, K.; Taesuwan, S.; Yaffe, R.; Caudill, M.A.; Golan, H.M. Prenatal Nutritional Intervention Reduces Autistic-Like Behavior Rates Among Mthfr-Deficient Mice. Front. Neurosci. 2019, 13, 383. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.J.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tancredi, D.J.; Tassone, F.; Hertz-Picciotto, I. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology 2011, 22, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yu, L.; Li, S.; Liu, J. Association Study of Polymorphisms in Genes Relevant to Vitamin B12 and Folate Metabolism with Childhood Autism Spectrum Disorder in a Han Chinese Population. Med. Sci. Monit. 2018, 24, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Fadila; Suman, P.; Kumar, P.; Omair, F. Clinical Relevance of Methylenetetrahydrofolate Reductase Genetic Testing in Autism: A Case Report of Successful Clinical Outcome. Cureus 2021, 13, e12586. [Google Scholar] [CrossRef]
- Boris, M.; Goldblatt, P.A.; Galanko, J.; James, S.J. Association of MTHFR Gene Variants with Autism. J. Am. Physicians Surg. 2004, 9, 3. [Google Scholar]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.; Lucock, M.; Stuart, J.; Fardell, S.; Baker, K.; Ng, X. Preliminary evidence for involvement of the folate gene polymorphism 19bp deletion-DHFR in occurrence of autism. Neurosci. Lett. 2007, 422, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Solehdin, F.; Cohen, I.L.; Gonzalez, M.G.; Jenkins, E.C.; Lewis, M.E.; Holden, J.J. Population- and family-based studies associate the MTHFR gene with idiopathic autism in simplex families. J. Autism Dev. Disord. 2011, 41, 938–944. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Tancredi, D.J.; Ozonoff, S.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tassone, F.; Hertz-Picciotto, I. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am. J. Clin. Nutr. 2012, 96, 80–89. [Google Scholar] [CrossRef]
- Guo, T.; Chen, H.; Liu, B.; Ji, W.; Yang, C. Methylenetetrahydrofolate reductase polymorphisms C677T and risk of autism in the Chinese Han population. Genet. Test. Mol. Biomark. 2012, 16, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ro, M.; Pyun, J.A.; Nam, M.; Bang, H.J.; Yang, J.W.; Choi, K.S.; Kim, S.K.; Chung, J.H.; Kwack, K. MTHFR 1298A>C is a risk factor for autism spectrum disorder in the Korean population. Psychiatry Res. 2014, 215, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Rai, V. Association of methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism with autism: Evidence of genetic susceptibility. Metab. Brain Dis. 2016, 31, 727–735. [Google Scholar] [CrossRef]
- El-Baz, F.; El-Aal, M.A.; Kamal, T.M.; Sadek, A.A.; Othman, A.A. Study of the C677T and 1298AC polymorphic genotypes of MTHFR Gene in autism spectrum disorder. Electron. Physician 2017, 9, 5287–5293. [Google Scholar] [CrossRef] [Green Version]
- El Shafae, M.; Sabry, J.H.; Behiry, E.G.; Elshahat, S.A.; Zaki, M.S.; Esmaiel, N.N. Association of cystathionine β synthase gene polymorphism with cognitive disorders in autistic children. J. Innov. Pharm. Biol. Sci. JIPBS 2017, 4, 5. [Google Scholar]
- Sadeghiyeh, T.; Dastgheib, S.A.; Mirzaee-Khoramabadi, K.; Morovati-Sharifabad, M.; Akbarian-Bafghi, M.J.; Poursharif, Z.; Mirjalili, S.R.; Neamatzadeh, H. Association of MTHFR 677C>T and 1298A>C polymorphisms with susceptibility to autism: A systematic review and meta-analysis. Asian J. Psychiatr. 2019, 46, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, S.; Shi, J.; Guo, Y.; Li, Z.; Cheng, Y.; Liu, Y. Association between MTHFR C677T/A1298C and susceptibility to autism spectrum disorders: A meta-analysis. BMC Pediatr. 2020, 20, 449. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.; Selhub, J.; Paul, L.; Ji, Y.; Wang, G.; Hong, X.; Zuckerman, B.; Fallin, M.D.; Wang, X. A prospective birth cohort study on cord blood folate subtypes and risk of autism spectrum disorder. Am. J. Clin. Nutr. 2020, 112, 1304–1317. [Google Scholar] [CrossRef]
- Banka, S.; Blom, H.J.; Walter, J.; Aziz, M.; Urquhart, J.; Clouthier, C.M.; Rice, G.I.; de Brouwer, A.P.; Hilton, E.; Vassallo, G.; et al. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am. J. Hum. Genet. 2011, 88, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef] [Green Version]
- Stanislawska-Sachadyn, A.; Brown, K.S.; Mitchell, L.E.; Woodside, J.V.; Young, I.S.; Scott, J.M.; Murray, L.; Boreham, C.A.; McNulty, H.; Strain, J.J.; et al. An insertion/deletion polymorphism of the dihydrofolate reductase (DHFR) gene is associated with serum and red blood cell folate concentrations in women. Hum. Genet. 2008, 123, 289–295. [Google Scholar] [CrossRef]
- Burdennyy, A.M.; Loginov, V.I.; Zavarykina, T.M.; Braga, E.A.; Kubatiev, A.A. The role of molecular genetic alterations in genes involved in folate and homocysteine metabolism in multifactorial diseases pathogenesis. Russ. J. Genet. 2017, 53, 528–541. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; Quadros, E.V. The basis for folinic acid treatment in neuro-psychiatric disorders. Biochimie 2016, 126, 79–90. [Google Scholar] [CrossRef]
- Chen, M.J.; Shimada, T.; Moulton, A.D.; Cline, A.; Humphries, R.K.; Maizel, J.; Nienhuis, A.W. The functional human dihydrofolate reductase gene. J. Biol. Chem. 1984, 259, 3933–3943. [Google Scholar] [CrossRef]
- Higgins, C.E.; Gross, S.S. Chapter 6—Tetrahydrobiopterin: An Essential Cofactor for Nitric Oxide Synthases and Amino Acid Hydroxylases. In Nitric Oxide, 2nd ed.; Ignarro, L.J., Ed.; Academic Press: San Diego, CA, USA, 2010; pp. 169–209. [Google Scholar] [CrossRef]
- Autism Genome Project, C.; Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Fellah, S.; Cheung, Y.T.; Scoggins, M.A.; Zou, P.; Sabin, N.D.; Pui, C.H.; Robison, L.L.; Hudson, M.M.; Ogg, R.J.; Krull, K.R. Brain Activity Associated With Attention Deficits Following Chemotherapy for Childhood Acute Lymphoblastic Leukemia. J. Natl. Cancer Inst. 2019, 111, 201–209. [Google Scholar] [CrossRef]
- Zajac-Spychala, O.; Pawlak, M.; Karmelita-Katulska, K.; Pilarczyk, J.; Jonczyk-Potoczna, K.; Przepiora, A.; Derwich, K.; Wachowiak, J. Anti-leukemic treatment-induced neurotoxicity in long-term survivors of childhood acute lymphoblastic leukemia: Impact of reduced central nervous system radiotherapy and intermediate- to high-dose methotrexate. Leuk. Lymphoma 2018, 59, 2342–2351. [Google Scholar] [CrossRef]
- Wiens, D.; DeSoto, M.C. Is High Folic Acid Intake a Risk Factor for Autism?—A Review. Brain Sci. 2017, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Neggers, Y. The Relationship between Folic Acid and Risk of Autism Spectrum Disorders. Healthcare 2014, 2, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Plumptre, L.; Masih, S.P.; Ly, A.; Aufreiter, S.; Sohn, K.J.; Croxford, R.; Lausman, A.Y.; Berger, H.; O’Connor, D.L.; Kim, Y.I. High concentrations of folate and unmetabolized folic acid in a cohort of pregnant Canadian women and umbilical cord blood. Am. J. Clin. Nutr. 2015, 102, 848–857. [Google Scholar] [CrossRef]
- Obeid, R.; Kirsch, S.H.; Dilmann, S.; Klein, C.; Eckert, R.; Geisel, J.; Herrmann, W. Folic acid causes higher prevalence of detectable unmetabolized folic acid in serum than B-complex: A randomized trial. Eur. J. Nutr. 2016, 55, 1021–1028. [Google Scholar] [CrossRef]
- Lubinsky, M. An epigenetic association of malformations, adverse reproductive outcomes, and fetal origins hypothesis related effects. J. Assist. Reprod. Genet. 2018, 35, 953–964. [Google Scholar] [CrossRef]
- Sulistyoningrum, D.; Green, T.; Palmer, D.; Sullivan, T.; Wood, S.; Makrides, M.; Skubisz, M.; Best, K.P. Study protocol for a randomised controlled trial evaluating the effect of folic acid supplementation beyond the first trimester on maternal plasma unmetabolised folic acid in late gestation. BMJ Open 2020, 10, e040416. [Google Scholar] [CrossRef]
- Kraus, J.P.; Oliveriusova, J.; Sokolova, J.; Kraus, E.; Vlcek, C.; de Franchis, R.; Maclean, K.N.; Bao, L.; Bukovská, G.; Patterson, D.; et al. The human cystathionine β-synthase (CBS) gene: Complete sequence, alternative splicing, and polymorphisms. Genomics 1998, 52, 312–324. [Google Scholar] [CrossRef]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of human cystathionine β-synthase: A unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, H.; Roh, H.; Kwon, Y. Causes of hyperhomocysteinemia and its pathological significance. Arch. Pharm. Res. 2018, 41, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.A.; Kozich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine β-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Menezo, Y.; Mares, P.; Cohen, M.; Brack, M.; Viville, S.; Elder, K. Autism, imprinting and epigenetic disorders: A metabolic syndrome linked to anomalies in homocysteine recycling starting in early life? J. Assist. Reprod. Genet. 2011, 28, 1143–1145. [Google Scholar] [CrossRef] [Green Version]
- Menezo, Y.J.; Elder, K.; Dale, B. Link Between Increased Prevalence of Autism Spectrum Disorder Syndromes and Oxidative Stress, DNA Methylation, and Imprinting: The Impact of the Environment. JAMA Pediatr. 2015, 169, 1066–1067. [Google Scholar] [CrossRef] [Green Version]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.M. Autism biomarkers: Challenges, pitfalls and possibilities. J. Autism Dev. Disord. 2015, 45, 1103–1113. [Google Scholar] [CrossRef]
- Tognazzo, S.; Gemmati, D.; Palazzo, A.; Catozzi, L.; Carandina, S.; Legnaro, A.; Tacconi, G.; Scapoli, G.L.; Zamboni, P. Prognostic role of factor XIII gene variants in nonhealing venous leg ulcers. J. Vasc. Surg. 2006, 44, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.V.; Vyas, V.; Montani, E.; Cartelli, D.; Parazzoli, D.; Oldani, A.; Zeri, G.; Orioli, E.; Gemmati, D.; Zamboni, P. Investigation of in vitro cytotoxicity of the redox state of ionic iron in neuroblastoma cells. J. Neurosci. Rural. Pract. 2012, 3, 301–310. [Google Scholar] [CrossRef]
- Golimbet, V.; Korovaitseva, G.; Abramova, L.; Kaleda, V. The 844ins68 polymorphism of the cystathionine β-synthase gene is associated with schizophrenia. Psychiatry Res. 2009, 170, 168–171. [Google Scholar] [CrossRef]
- Lechpammer, M.; Tran, Y.P.; Wintermark, P.; Martinez-Cerdeno, V.; Krishnan, V.V.; Ahmed, W.; Berman, R.F.; Jensen, F.E.; Nudler, E.; Zagzag, D. Upregulation of cystathionine β-synthase and p70S6K/S6 in neonatal hypoxic ischemic brain injury. Brain Pathol. 2017, 27, 449–458. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Adesina, I.; McGillivray, J.; Rinehart, N.J. Sex differences in neurodevelopmental disorders. Curr. Opin. Neurol. 2019, 32, 622–626. [Google Scholar] [CrossRef]
- Piccini, P.; Montagnani, C.; de Martino, M. Gender disparity in pediatrics: A review of the current literature. Ital. J. Pediatr. 2018, 44, 1. [Google Scholar] [CrossRef]
- Braunstein, V.L.; Peniston, N.; Perelman, A.; Cassano, M.C. The inclusion of fathers in investigations of autistic spectrum disorders. Res. Autism Spectr. Disord. 2013, 7, 8. [Google Scholar] [CrossRef]
- Volkmar, F.R.; Szatmari, P.; Sparrow, S.S. Sex differences in pervasive developmental disorders. J. Autism Dev. Disord. 1993, 23, 579–591. [Google Scholar] [CrossRef]
- Szatmari, P.; Liu, X.Q.; Goldberg, J.; Zwaigenbaum, L.; Paterson, A.D.; Woodbury-Smith, M.; Georgiades, S.; Duku, E.; Thompson, A. Sex differences in repetitive stereotyped behaviors in autism: Implications for genetic liability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 5–12. [Google Scholar] [CrossRef]
- Merikangas, A.K.; Almasy, L. Using the tools of genetic epidemiology to understand sex differences in neuropsychiatric disorders. Genes Brain Behav. 2020, 19, e12660. [Google Scholar] [CrossRef]
- Szatmari, P.; Georgiades, S.; Duku, E.; Bennett, T.A.; Bryson, S.; Fombonne, E.; Mirenda, P.; Roberts, W.; Smith, I.M.; Vaillancourt, T.; et al. Developmental trajectories of symptom severity and adaptive functioning in an inception cohort of preschool children with autism spectrum disorder. JAMA Psychiatry 2015, 72, 276–283. [Google Scholar] [CrossRef]
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009, 65, 591–598. [Google Scholar] [CrossRef]
- Geary, D.C. Autism in the broader context of cognitive sex differences. Proc. Natl. Acad. Sci. USA 2018, 115, 12089–12091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zander, E.; Sturm, H.; Bolte, S. The added value of the combined use of the Autism Diagnostic Interview-Revised and the Autism Diagnostic Observation Schedule: Diagnostic validity in a clinical Swedish sample of toddlers and young preschoolers. Autism 2015, 19, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.E. Gender and coping: The parents of children with high functioning autism. Soc. Sci. Med. 2003, 56, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Lombardo, M.V.; Auyeung, B.; Chakrabarti, B.; Baron-Cohen, S. Sex/gender differences and autism: Setting the scene for future research. J. Am. Acad. Child Adolesc. Psychiatry 2015, 54, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Bartley, J.J. An update on autism: Science, gender, and the law. Gend. Med. 2006, 3, 73–78. [Google Scholar] [CrossRef]
- Burghardt, K.J.; Pilsner, J.R.; Bly, M.J.; Ellingrod, V.L. DNA methylation in schizophrenia subjects: Gender and MTHFR 677C/T genotype differences. Epigenomics 2012, 4, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Blumkin, E.; Levav-Rabkin, T.; Melamed, O.; Galron, D.; Golan, H.M. Gender-specific effect of Mthfr genotype and neonatal vigabatrin interaction on synaptic proteins in mouse cortex. Neuropsychopharmacology 2011, 36, 1714–1728. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Xavier, M.J.; Roman, S.D.; Aitken, R.J.; Nixon, B. Transgenerational inheritance: How impacts to the epigenetic and genetic information of parents affect offspring health. Hum. Reprod. Update 2019, 25, 518–540. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Hong, K.; Liu, R.; Inoue, A.; Shen, L.; Zhang, K.; Zhang, Y. Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during germ cell reprogramming. Cell Res. 2013, 23, 329–339. [Google Scholar] [CrossRef]
- Mastrototaro, G.; Sessa, A. Chapter 9—Emerging Role of Epigenetics in Human Neurodevelopmental Disorders. In Epigenetics in Human Disease, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 6, pp. 269–304. [Google Scholar]
- Gegenhuber, B.; Tollkuhn, J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes 2019, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P.; Sharp, A.J. Screening for rare epigenetic variations in autism and schizophrenia. Hum. Mutat. 2019, 40, 952–961. [Google Scholar] [CrossRef]
- Sabit, H.; Tombuloglu, H.; Rehman, S.; Almandil, N.B.; Cevik, E.; Abdel-Ghany, S.; Rashwan, S.; Abasiyanik, M.F.; Yee Waye, M.M. Gut microbiota metabolites in autistic children: An epigenetic perspective. Heliyon 2021, 7, e06105. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Hu, V.W.; Hong, Y.; Xu, M.; Shu, H.T. Altered DNA methylation in a severe subtype of idiopathic autism: Evidence for sex differences in affected metabolic pathways. Autism 2020, 25, 887–910. [Google Scholar] [CrossRef]
- Siu, M.T.; Goodman, S.J.; Yellan, I.; Butcher, D.T.; Jangjoo, M.; Grafodatskaya, D.; Rajendram, R.; Lou, Y.; Zhang, R.; Zhao, C.; et al. DNA Methylation of the Oxytocin Receptor Across Neurodevelopmental Disorders. J. Autism Dev. Disord. 2021. [Google Scholar] [CrossRef]
- Banik, A.; Kandilya, D.; Ramya, S.; Stunkel, W.; Chong, Y.S.; Dheen, S.T. Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring. Genes 2017, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Kundakovic, M.; Jaric, I. The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes 2017, 8, 104. [Google Scholar] [CrossRef]
- Abdalla, H.; Yoshizawa, Y.; Hochi, S. Active demethylation of paternal genome in mammalian zygotes. J. Reprod. Dev. 2009, 55, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Prasoona, K.R.; Sunitha, T.; Srinadh, B.; Muni Kumari, T.; Jyothy, A. Maternal association and influence of DHFR 19 bp deletion variant predisposes foetus to anencephaly susceptibility: A family-based triad study. Biomarkers 2018, 23, 640–646. [Google Scholar] [CrossRef]
- Ahmed, O.G.; Shehata, G.A.; Ali, R.M.; Makboul, R.; Abd Allah, E.S.H.; Abd El-Rady, N.M. Folic acid ameliorates neonatal isolation-induced autistic like behaviors in rats: Epigenetic modifications of BDNF and GFAP promotors’. Appl. Physiol. Nutr. Metab. 2021. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; de Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.; Bok, L.A.; et al. Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef]
- Van Winkle, L.J.; Ryznar, R. One-Carbon Metabolism Regulates Embryonic Stem Cell Fate Through Epigenetic DNA and Histone Modifications: Implications for Transgenerational Metabolic Disorders in Adults. Front. Cell Dev. Biol. 2019, 7, 300. [Google Scholar] [CrossRef] [Green Version]
- Mitra, I.; Huang, B.; Mousavi, N.; Ma, N.; Lamkin, M.; Yanicky, R.; Shleizer-Burko, S.; Lohmueller, K.E.; Gymrek, M. Patterns of de novo tandem repeat mutations and their role in autism. Nature 2021, 589, 246–250. [Google Scholar] [CrossRef]
- Searles Quick, V.B.; Wang, B.; State, M.W. Leveraging large genomic datasets to illuminate the pathobiology of autism spectrum disorders. Neuropsychopharmacology 2021, 46, 55–69. [Google Scholar] [CrossRef]
- Kim, N.; Kim, K.H.; Lim, W.J.; Kim, J.; Kim, S.A.; Yoo, H.J. Whole Exome Sequencing Identifies Novel De Novo Variants Interacting with Six Gene Networks in Autism Spectrum Disorder. Genes 2020, 12, 1. [Google Scholar] [CrossRef]
- Emberti Gialloreti, L.; Enea, R.; Di Micco, V.; Di Giovanni, D.; Curatolo, P. Clustering Analysis Supports the Detection of Biological Processes Related to Autism Spectrum Disorder. Genes 2020, 11, 1476. [Google Scholar] [CrossRef]
- Mesleh, A.G.; Abdulla, S.A.; El-Agnaf, O. Paving the Way toward Personalized Medicine: Current Advances and Challenges in Multi-OMICS Approach in Autism Spectrum Disorder for Biomarkers Discovery and Patient Stratification. J. Pers. Med. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, L.R.; Cisternas, C.D.; Forger, N.G. Does Gender Leave an Epigenetic Imprint on the Brain? Front. Neurosci. 2019, 13, 173. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
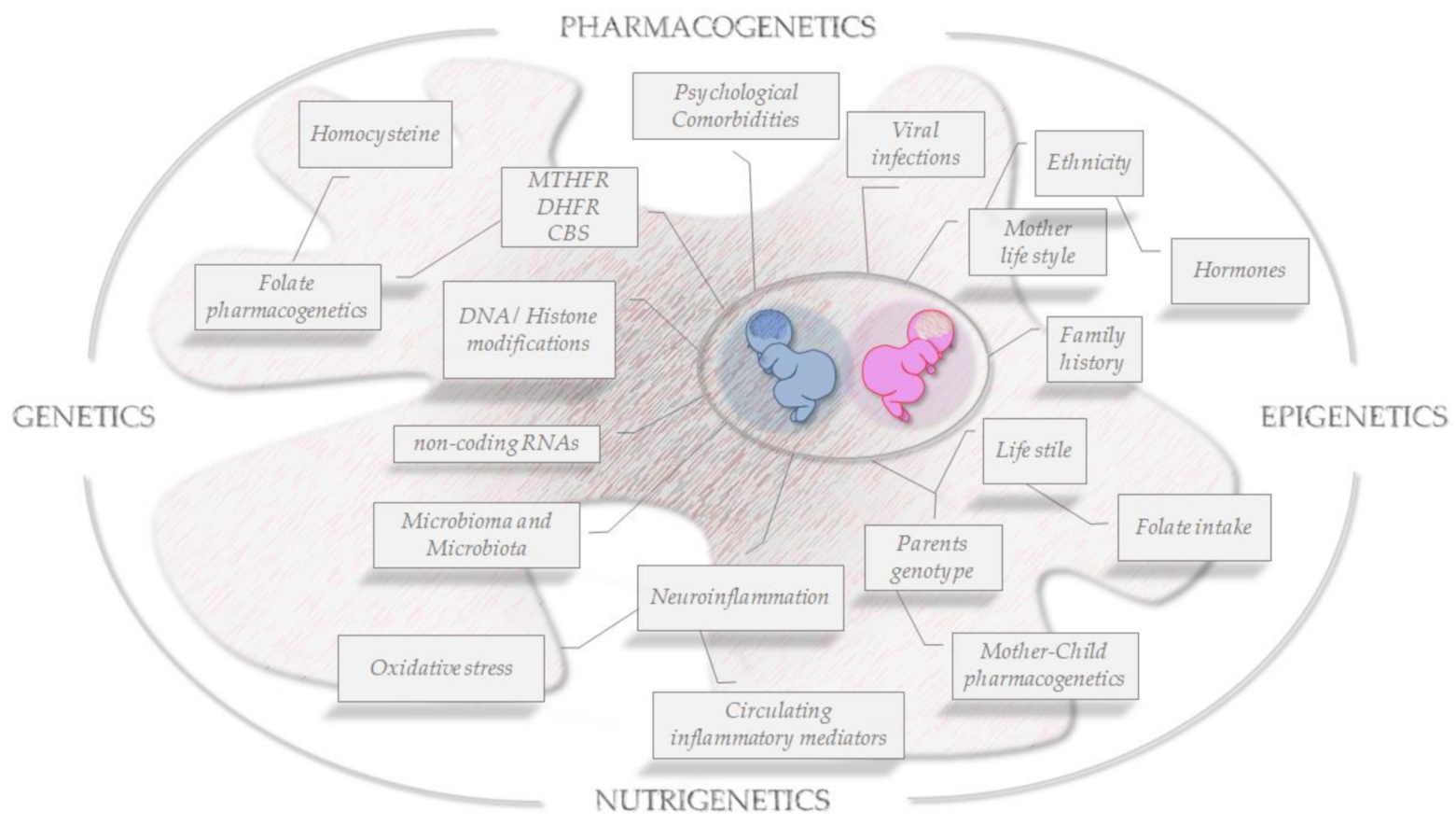{kind=link}
| Gene | Key Findings | Genotype/Allele | Ref |
|---|---|---|---|
| MTHFR | • Increased frequency of MTHFR C677T in ASD • Unexpected high frequency of the normal MTHFR 1298AA genotype in ASD • Combined MTHFR 677CT/1298AC haplotype more prevalent in ASD | 677 CT/TT 1298 AA | [84] |
| MTHFR | • Circulating methionine and SAM/SAH ratio are significantly decreased in ASD • Circulating cysteine, GSH, and GSH/GSSG ratio are significantly decreased in ASD • Disease association with MTHFR C677T and A1298C | 677 CT+TT 1298/677 AC/CT combined with RFC 80GA | [85] |
| DHFR | • DHFR 19bp ins/del is a risk factor for ASD independently from and in association with folate polymorphisms • DHFR 19bp ins/del combined with MTHFR C677T and A1298C | 19bp del 19bp del+677T+1298C | [86] |
| MTHFR | • MTHFR C677T emerges as primary ASD risk factor • MTHFR A1298C emerges as additive risk factor for ASD in combination to C677T | 677 T 677 T + 1298C | [72] |
| MTHFR | • High frequency of MTHFR 677 T-allele and TT-genotype, 677/1298 T/A and TT/AA haplotypes in ASD • Preferential parental transmission of 677 T- and 1298 A-allele or 677/1298 T/A haplotypes in affected offspring | 677 T 1298 AA | [87] |
| MTHFRCBS | • Periconceptional vitamins intake reduces the risk of having ASD children in genetically susceptible mothers/children dyad • Higher ASD risk in mother MTHFR 677TT, CBS rs234715 GT+TT with child COMT 472 AA genotypes • Higher ASD risk in mothers also carrying other one-carbon metabolism gene variants | 677 TT combined with other one-carbon gene variants, both in mother and child | [81] |
| MTHFR | • Lower ASD risk associated to folic acid supplement strongest in MTHFR C677T carriers (mothers/children) | 677 CT+TT | [88] |
| MTHFR | • High frequency of MTHFR 677TT in ASD children • Over-activity significantly associated to MTHFR 677TT genotype (Stratification by Autism Diagnostic Interview) | 677 TT | [89] |
| MTHFR | • Meta-analysis: eight case-control studies included • Higher ASD risk to MTHFR C677T polymorphism (all comparison models) • Lower ASD risk to MTHFR A1298C polymorphism (recessive model) • ASD association to MTHFR C677T polymorphism (only in countries without food fortification) | 677 CT+TT 1298 CC | [71] |
| MTHFR | • Associations to ASD with MTHFR A1298C • Higher ASD risk to MTHFR 677CT/1298AC combined genotype • No significant associations in females | 1298 AC+CC 677/1298 CT/AC | [90] |
| MTHFR | • Meta-analysis: thirteen studies included (9 on Caucasians, 4 on Asians) • Significant association between ASD and MTHFR C677T polymorphism | 677 CT+TT 677 TT 677 T | [91] |
| MTHFR | • Higher MTHFR A1298C frequency in ASD (AC: 41.9%; CC: 35.5%) • Higher MTHFR C677T frequency in ASD (CT: 48.4%; TT 12.9%) • Heterozygosity was equally detected (46.2%) among patients with severe autism | 677 CT+TT 677 T 1298 AC+CC 1298 C | [92] |
| CBS | • Higher CBS C699T frequencies distributions (TT and CT+TT) in ASD patients • Lower CBS C699T frequency associated with sleep and GIT disorders • No significant association between CBS genotypes and severity of ASD | 699 CT+TT 699 TT | [93] |
| MTHFR | • Meta-analysis: 25 case-control studies on MTHFR (C677T, 18 studies) (A1298C, 7 studies) • Higher MTHFR C677T frequency in ASD • No overall association between MTHFR A1298C and ASD risk MTHFR A1298C significantly associated only in Caucasians | 677 CT+TT 677 TT 677 T 1298 CC 1298 C | [94] |
| MTHFR | • Meta-analysis: 15 studies • Higher ASD risk to MTHFR C677T polymorphism (all comparison models) • No association between MTHFR A1298C and ASD (all comparison models) | 677 CT+TT 677 TT 677 T 677/1298 T/C | [95] |
| DHFR | • Positive association (not adjusted) between cord total folate and UMFA also after DHFR genotype stratification (limited to Black children) | 19bp del/del | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisato, V.; Silva, J.A.; Longo, G.; Gallo, I.; Singh, A.V.; Milani, D.; Gemmati, D. Genetics and Epigenetics of One-Carbon Metabolism Pathway in Autism Spectrum Disorder: A Sex-Specific Brain Epigenome? Genes 2021, 12, 782. https://doi.org/10.3390/genes12050782
Tisato V, Silva JA, Longo G, Gallo I, Singh AV, Milani D, Gemmati D. Genetics and Epigenetics of One-Carbon Metabolism Pathway in Autism Spectrum Disorder: A Sex-Specific Brain Epigenome? Genes. 2021; 12(5):782. https://doi.org/10.3390/genes12050782
Chicago/Turabian StyleTisato, Veronica, Juliana A. Silva, Giovanna Longo, Ines Gallo, Ajay V. Singh, Daniela Milani, and Donato Gemmati. 2021. "Genetics and Epigenetics of One-Carbon Metabolism Pathway in Autism Spectrum Disorder: A Sex-Specific Brain Epigenome?" Genes 12, no. 5: 782. https://doi.org/10.3390/genes12050782
APA StyleTisato, V., Silva, J. A., Longo, G., Gallo, I., Singh, A. V., Milani, D., & Gemmati, D. (2021). Genetics and Epigenetics of One-Carbon Metabolism Pathway in Autism Spectrum Disorder: A Sex-Specific Brain Epigenome? Genes, 12(5), 782. https://doi.org/10.3390/genes12050782