
Clinical and Molecular Features of Skin Malignancies in Muir-Torre Syndrome


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Introduction
2.2. Mutational Analysis of Tumor Samples
3. Results
3.1. Demographic Data
3.2. Medical History
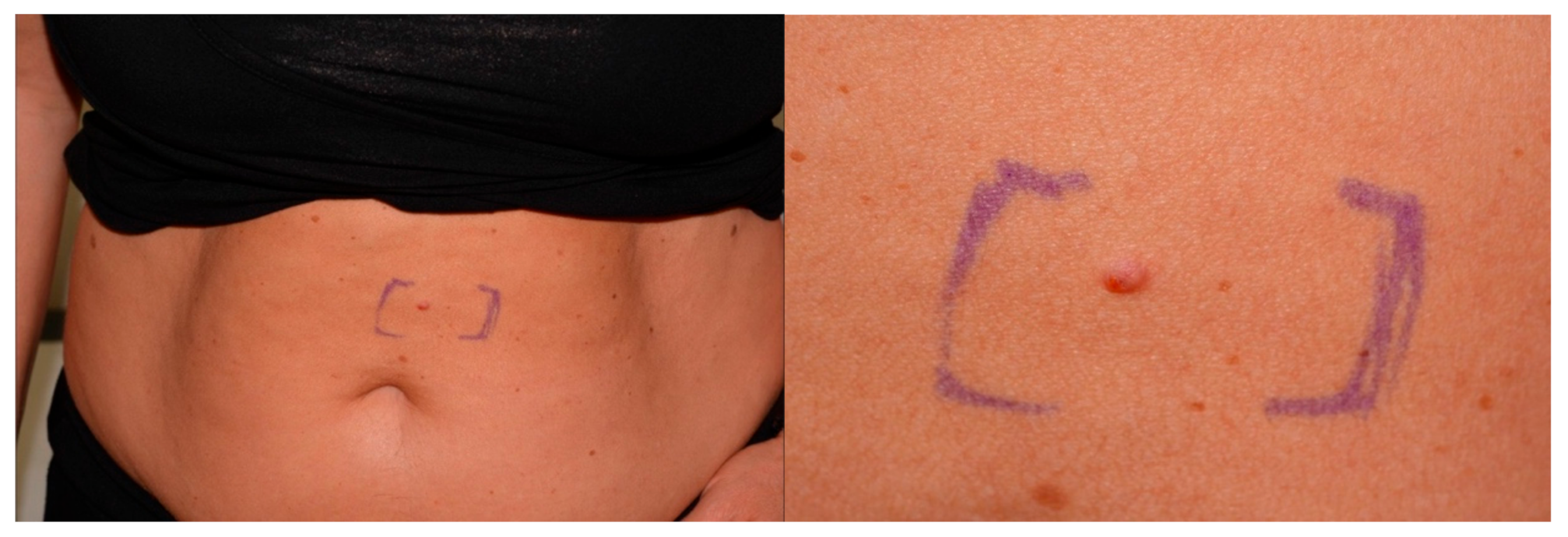
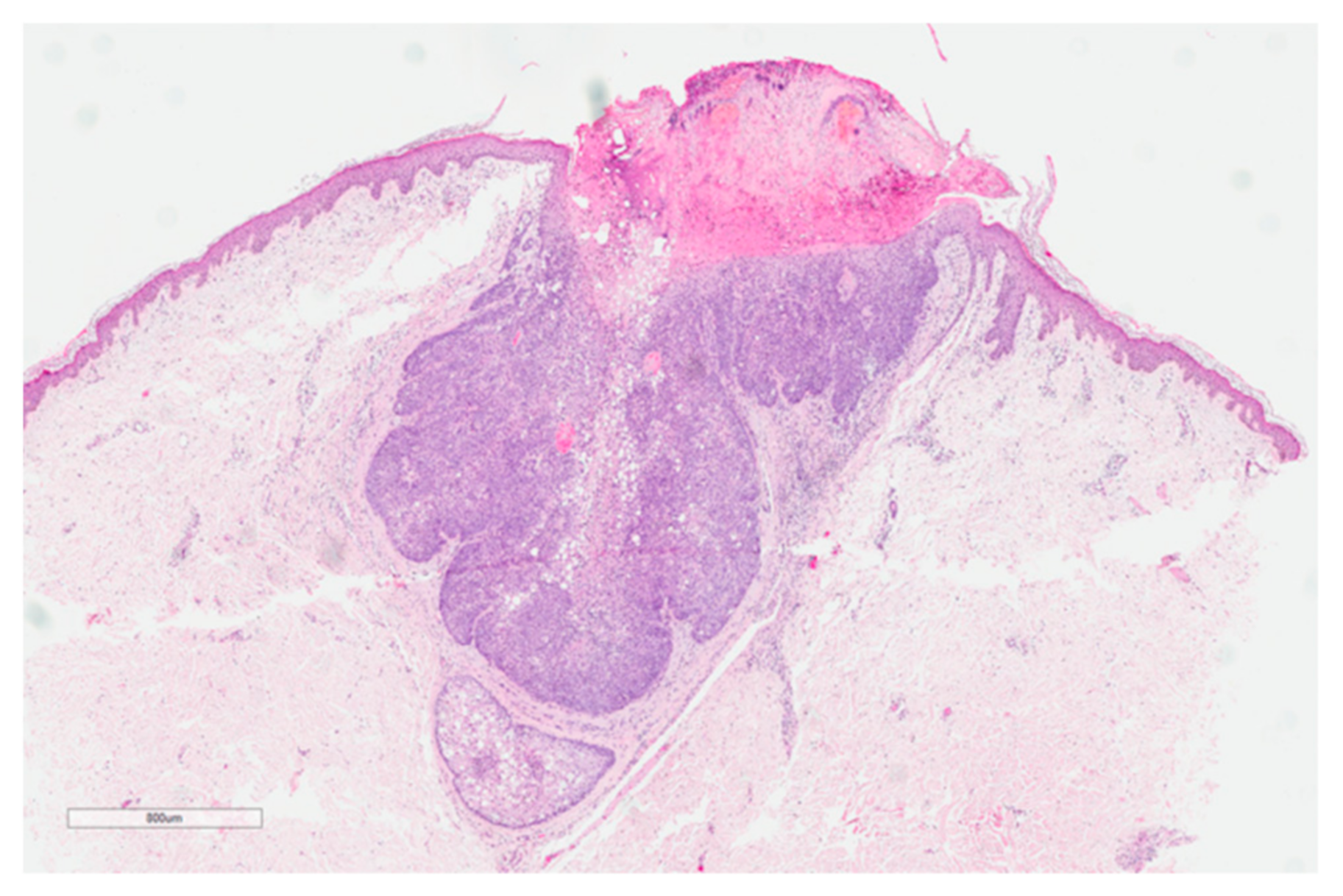
3.3. Sebaceous Lesions and Other Cutaneous Tumors
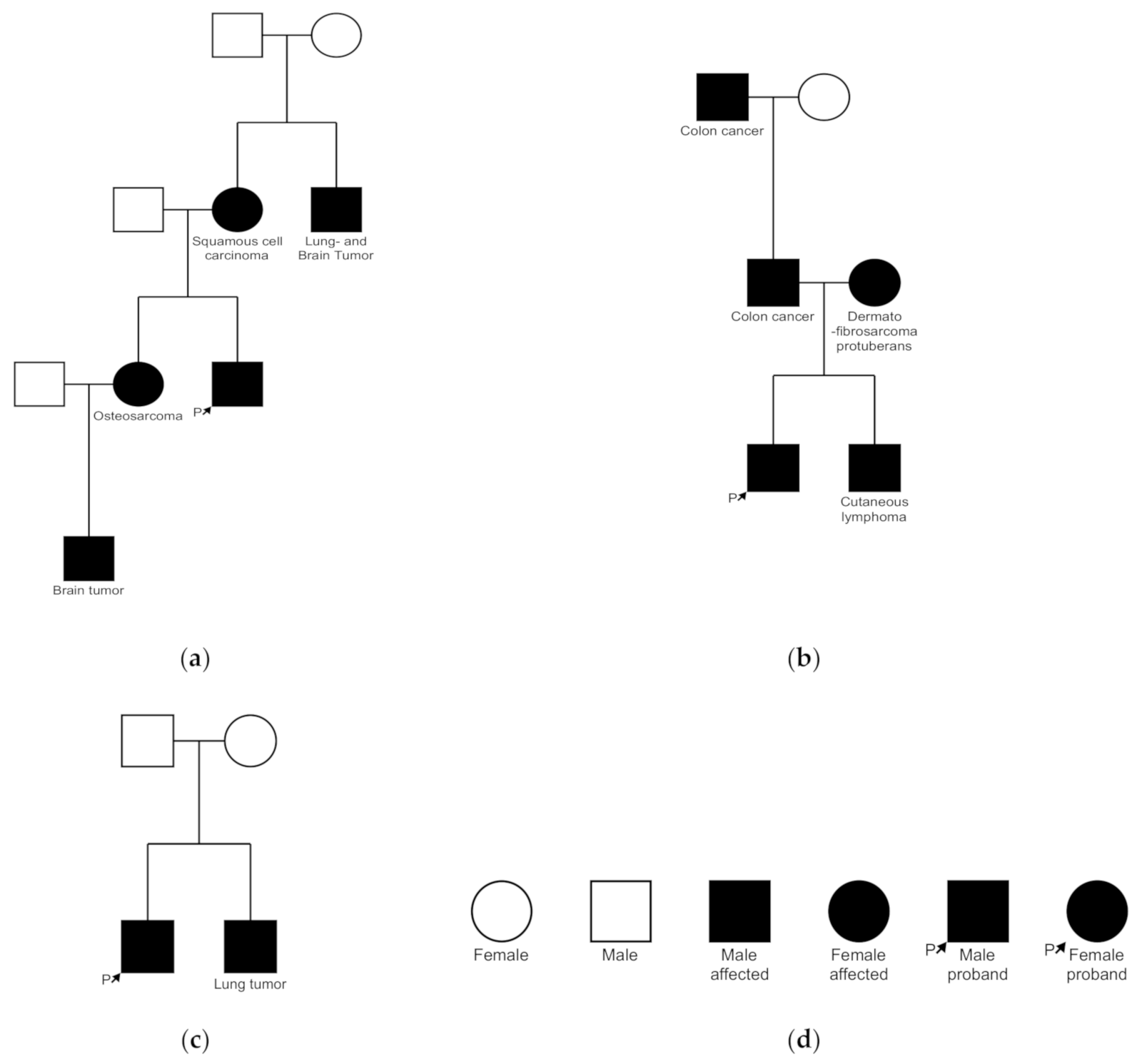
3.4. Family History
3.5. Tumor Mutations
4. Discussion
4.1. Introduction
4.2. Demographic Data
4.3. Medical History
4.4. Family History
4.5. Mutational Analysis
4.6. Diagnostic Criteria
4.7. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joly, M.-O.; Attignon, V.; Saurin, J.-C.; Desseigne, F.; Leroux, D.; Martin-Denavit, T.; Giraud, S.; Bonnet-Dupeyron, M.-N.; Faivre, L.; Auclair, J.; et al. Somatic MMR gene mutations as a cause for MSI-H sebaceous neoplasms in Muir–Torre syndrome-like patients. Hum. Mutat. 2015, 36, 292–295. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Geurts van Kessel, A.; Hoogerbrugge, N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam. Cancer 2013, 12, 169–174. [Google Scholar] [CrossRef]
- Muir, E.G.; Bell, A.J.; Barlow, K.A. Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. Br. J. Surg. 1967, 54, 191–195. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; French, A.J.; Roche, P.C.; Cunningham, J.M.; Tester, D.J.; Lindor, N.M.; Moslein, G.; Baker, S.M.; Liskay, R.M.; Burgart, L.J.; et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996, 56, 4836–4840. [Google Scholar]
- Entius, M.M.; Keller, J.J.; Drillenburg, P.; Kuypers, K.C.; Giardiello, F.M.; Offerhaus, G.J. Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin. Cancer Res. 2000, 6, 1784–1789. [Google Scholar]
- Steinke, V.; Engel, C.; Büttner, R.; Schackert, H.K.; Schmiegel, W.H.; Propping, P. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. Dtsch. Arztebl. Int. 2013, 110, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008, 135, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Baglietto, L.; Lindor, N.M.; Dowty, J.G.; White, D.M.; Wagner, A.; Gomez Garcia, E.B.; Vriends, A.H.; Cartwright, N.R.; Barnetson, R.A.; Farrington, S.M.; et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J. Natl. Cancer Inst. 2010, 102, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Le, S.; Ansari, U.; Mumtaz, A.; Malik, K.; Patel, P.; Doyle, A.; Khachemoune, A. Lynch Syndrome and Muir-Torre Syndrome: An update and review on the genetics, epidemiology, and management of two related disorders. Dermatol. Online J. 2017, 23. [Google Scholar]
- Nelson, B.R.; Hamlet, K.R.; Gillard, M.; Railan, D.; Johnson, T.M. Sebaceous carcinoma. J. Am. Acad. Dermatol. 1995, 33, 1–15. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Burris, C.K.H.; Rodriguez, M.E.; Raven, M.L.; Reddy, D.N.; Xu, Y.G.; Wiggs, J.L.; Potter, H.D.; Albert, D.M. Muir-Torre Syndrome: The importance of a detailed family history. Case Rep. Ophthalmol. 2019, 10, 180–185. [Google Scholar] [CrossRef]
- Hatta, N.; Takata, A.; Ishizawa, S.; Niida, Y. Family with MSH2 mutation presenting with keratoacanthoma and precancerous skin lesions. J. Dermatol. 2015, 42, 1087–1090. [Google Scholar] [CrossRef]
- Kacerovska, D.; Cerna, K.; Martinek, P.; Grossmann, P.; Michal, M.; Ricar, J.; Kazakov, D.V. MSH6 mutation in a family affected by Muir-Torre syndrome. Am. J. Dermatopathol. 2012, 34, 648–652. [Google Scholar] [CrossRef]
- Abbas, O.; Mahalingam, M. Cutaneous sebaceous neoplasms as markers of Muir-Torre syndrome: A diagnostic algorithm. J. Cutan. Pathol. 2009, 36, 613–619. [Google Scholar] [CrossRef]
- Gay, J.T.; Troxell, T.; Gross, G.P. Muir-Torre Syndrome; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Akhtar, S.; Oza, K.K.; Khan, S.A.; Wright, J. Muir-Torre syndrome: Case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J. Am. Acad. Dermatol. 1999, 41, 681–686. [Google Scholar] [CrossRef]
- Cohen, P.R.; Kohn, S.R.; Kurzrock, R. Association of sebaceous gland tumors and internal malignancy: The Muir-Torre syndrome. Am. J. Med. 1991, 90, 606–613. [Google Scholar] [CrossRef]
- Gray, S.E.; Kay, E.W.; Leader, M.; Mabruk, M.J. Enhanced detection of microsatellite instability and mismatch repair gene expression in cutaneous squamous cell carcinomas. Mol. Diagn. Ther. 2006, 10, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kientz, C.; Joly, M.-O.; Faivre, L.; Clemenson, A.; Dalac, S.; Lepage, C.; Chapusot, C.; Jacquot, C.; Schiappa, R.; Lebrun, M. A case report of Muir-Torre syndrome in a woman with breast cancer and MSI-Low skin squamous cell carcinoma. Hered. Cancer Clin. Pract. 2017, 15, 6. [Google Scholar] [CrossRef]
- Ponti, G.; Losi, L.; Pellacani, G.; Wannesson, L.; Cesinaro, A.M.; Venesio, T.; Petti, C.; Seidenari, S. Malignant melanoma in patients with hereditary nonpolyposis colorectal cancer. Br. J. Dermatol. 2008, 159, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Swale, V.J.; Bataille, V.A.; Quinn, A.G.; Ghali, L.; Patel, S.V.; Dove-Edwin, I.; Cerio, R.; McGregor, J.M. An association between sebaceous carcinoma and microsatellite instability in immunosuppressed organ transplant recipients. J. Investig. Dermatol. 2001, 116, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Griffard, E.A.; McCoppin, H.H.; Wieberg, J.; Feldman, M. The cutaneous effects of post-transplant immunosuppression with cyclosporine in Muir-Torre syndrome. J. Am. Acad. Dermatol. 2011, 64, e86–e87. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Riegert-Johnson, D.L.; Thomas, B.C.; Thomas, C.S.; Heckman, M.G.; Krishna, M.; DiCaudo, D.J.; Bridges, A.G.; Hunt, K.S.; Rumilla, K.M.; et al. Screening for Muir-Torre syndrome using mismatch repair protein immunohistochemistry of sebaceous neoplasms. J. Genet. Couns. 2013, 22, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.L.; Chao, C.F.; Ding, D.C.; Yu, C.P.; Chang, C.C.; Lai, H.C.; Yu, M.H.; Liu, H.S.; Chu, T.Y. Multiple epithelial and nonepithelial tumors in hereditary nonpolyposis colorectal cancer: Characterization of germline and somatic mutations of the MSH2 gene and heterogeneity of replication error phenotypes. Cancer Genet. Cytogenet. 2004, 153, 108–114. [Google Scholar] [CrossRef]
- Lewis, D.J.; Duvic, M. A possible association between mycosis fungoides and Muir-Torre syndrome: Two disorders with microsatellite instability. JAAD Case Rep. 2017, 3, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarisbrick, J.J.; Mitchell, T.J.; Calonje, E.; Orchard, G.; Russell-Jones, R.; Whittaker, S.J. Microsatellite instability is associated with hypermethylation of the hMLH1 gene and reduced gene expression in mycosis fungoides. J. Investig. Dermatol. 2003, 121, 894–901. [Google Scholar] [CrossRef]
- Kuwabara, K.; Suzuki, O.; Chika, N.; Kumamoto, K.; Minabe, T.; Fukuda, T.; Arai, E.; Tamaru, J.I.; Akagi, K.; Eguchi, H.; et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn. J. Clin. Oncol. 2018, 48, 514–521. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The varied roles of notch in cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [Green Version]
- Dotto, G.P. Notch tumor suppressor function. Oncogene 2008, 27, 5115–5123. [Google Scholar] [CrossRef] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. Notch signaling in leukemia. Annu. Rev. Pathol. 2008, 3, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Selfridge, J.E.; Wang, J.; Zhao, Y.; Cui, J.; Guda, K.; Wang, Z.; Zhu, Y. Mutations in TP53, ZNF750, and RB1 typify ocular sebaceous carcinoma. J. Genet. Genom. 2019, 46, 315–318. [Google Scholar] [CrossRef]
- Harvey, N.T.; Tabone, T.; Erber, W.; Wood, B.A. Circumscribed sebaceous neoplasms: A morphological, immunohistochemical and molecular analysis. Pathology 2016, 48, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, M.T.; Singh, R.R.; Seviour, E.G.; Curry, J.L.; Hudgens, C.W.; Bell, D.; Wimmer, D.A.; Ning, J.; Czerniak, B.A.; Zhang, L.; et al. Next-generation sequencing identifies high frequency of mutations in potentially clinically actionable genes in sebaceous carcinoma. J. Pathol. 2016, 240, 84–95. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 Mutations are common in desmoplastic melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef] [Green Version]
- Vaassen, P.; Durr, N.; Rohrig, A.; Willing, R.; Rosenbaum, T. Trametinib induces neurofibroma shrinkage and enables surgery. Neuropediatrics 2019, 50, 300–303. [Google Scholar] [CrossRef]
- Tryggvason, G.; Bayon, R.; Pagedar, N.A. Epidemiology of sebaceous carcinoma of the head and neck: Implications for lymph node management. Head Neck 2012, 34, 1765–1768. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.A. Heritable colorectal cancer syndromes: Recognition and preventive management. Gastroenterol. Clin. N. Am. 2002, 31, 1107–1131. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Total Number of Patients | 7 |
|---|---|
| Male | 5 |
| Female | 2 |
| Age (range) | 61 (45–75) |
| Localization | |
| Non- head and neck | 7 |
| Head and neck | 4 |
| Tumor type (total number) | 11 |
| Sebaceous adenoma | 2 |
| Sebaceoma | 1 |
| Solid-cystic sebaceous neoplasm | 1 |
| Sebaceous carcinoma | 4 |
| Tumor with sebaceous differentiation | 1 |
| Squamous cell carcinoma | 1 |
| Melanoma | 1 |
| Patient | Sebaceous Tumor (Age at Diagnosis) | Visceral Tumor (Age at Diagnosis) | Family History | Localization |
|---|---|---|---|---|
| 1 | 1. Sebaceous carcinoma (63) 2. Tumor with sebaceous differentiation (63) 3. Sebaceous adenoma (67) | Caecal carcinoma (44) Bladder carcinoma (63) Adenocarcinoma sigma (67) | Sister: Osteosarcoma Uncle (mother’s side): Lung- and brain tumor Sisters’ son: Brain tumor | 1. Thorax 2. Forehead 3. Back |
| 2 | Sebaceoma (55) | - | Brother: Cutaneous lymphoma Mother: Dermatofibrosarcoma Father and grandfather: Colorectal carcinoma | Abdomen |
| 3 | Sebaceous carcinoma (63) | Renal cell carcinoma (58) | - | Hip |
| 4 | Solid-cystic sebaceous tumor (62) | - | - | Back |
| 5 | Sebaceous carcinoma (65) | - | - | Back |
| 6 | Sebaceous carcinoma (75) | - | - | Back |
| 7 | Sebaceous adenoma (72) | Small lymphocytic lymphoma | Brother: Lung tumor | Cheek |
| Mutated Gene | MSH2 | MLH1 | NOTCH1 | NOTCH2 | TP53 |
|---|---|---|---|---|---|
| Total number (%) | 6 (86%) | 1 (14%) | 3 (43%) | 2 (29%) | 2 (29%) |
| Histological type | |||||
| SC | 4 | - | 2 | 2 | 1 |
| SCS | 1 | - | 1 | - | 1 |
| SE | 1 | - | - | - | - |
| SA | - | 1 | - | - | - |
| Localization | |||||
| Non-head and neck | 5 | 1 | 2 | 1 | 2 |
| Head and neck | 1 | - | 1 | 1 | - |
| P | Sex (m/f) | Tumor Type (Tumor Cell Content) | Locus | Genes | % Frequency | Amino Acid Change | Coverage | Exon | Transcript | Coding | ACMG Class |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | m | SC (80%) | chr1:120462204 | NOTCH2 | 42 | p.(Arg1838*) | 2000 | 31 | NM_024408.3 | c.5512C>T | Pathogenic (class 5) |
| chr5:67591079 | PIK3R1 | 46 | p.(Glu558fs) | 1992 | 13 | NM_181523.2 | c.1674_1675del | Uncertain significance (class 3) | |||
| chr9:139404265 | NOTCH1 | 45 | p.(Cys963*) | 1985 | 18 | NM_017617.4 | c.2889C>A | Likely pathogenic (class 4) | |||
| chr9:139405616 | NOTCH1 | 44 | p.(Thr859fs) | 1923 | 16 | NM_017617.4 | c.2574_2575ins | Likely pathogenic (class 4) | |||
| TDS (70%) | chr1:120491661 | NOTCH2 | 41 | p.(Ser856fs) | 1623 | 16 | NM_024408.3 | c.2566_2567del | Likely pathogenic (class 4) | ||
| chr3:37090403 | MLH1 | 43 | p.(Trp666*) | 2000 | 18 | NM_000249.3 | c.1998G>A | Pathogenic (class 5) | |||
| chr3:47103763 | SETD2 | 40 | p.(Asp2064fs) | 1043 | 14 | NM_014159.6 | c.6181_6182del | Uncertain significance (class 3) | |||
| chr9:139397730 | NOTCH1 | 39 | p.(Gln1691*) | 810 | 27 | NM_017617.4 | c.5071C>T | Pathogenic (class 5) | |||
| chr10:123279674 | FGFR2 | 35 | p.(Pro253His) | 1677 | 7 | NM_000141.4 | c.758C>A | Likely pathogenic (class 4) | |||
| chr16:3646385 | SLX4 | 5 | p.(Gln565*) | 519 | 8 | NM_032444.3 | c.1693C>T | Pathogenic (class 5) | |||
| M (50%) | chr16:2135234 | TSC2 | 5 | p.(Gln1525*) | 366 | 36 | NM_000548.4 | c.4573C>T | Pathogenic (class 5) | ||
| SA (80%) | chr1:120464352 | NOTCH2 | 9 | p.(Gln1765fs) | 1939 | 29 | NM_024408.3 | c.5293delC | Likely pathogenic (class 4) | ||
| SCC (70%) | chr9:139417303 | NOTCH1 | 13 | p.(Pro247fs) | 1007 | 4 | NM_017617.4 | c.740_741ins | Likely pathogenic (class 4) | ||
| chr4:153249385 | FBXW7 | 22 | p.(Arg465Cys) | 2000 | 9 | NM_033632.3 | c.1393C>T | Likely pathogenic (class 4) | |||
| chr17:37687555 | CDK12 | 27 | p.(Gly1487*) | 2000 | 14 | NM_016507.3 | c.4459G>T | Uncertain significance (class 3) | |||
| chr17:37879658 | ERBB2 | 30 | p.(Arg678Gln) | 1999 | 17 | NM_004448.3 | c.2033G>A | Uncertain significance (class 3) | |||
| 2 | f | SE (70%) | chr2:47698134 | MSH2 | 42 | p.(Asn566fs) | 1907 | 11 | NM_000251.2 | c.1697del | Pathogenic (class 5) |
| chr7:55241677 | EGFR | 16 | p.(Glu709Lys) | 1997 | 18 | NM_005228.4 | c.2125G>A | Uncertain significance (class 3) | |||
| 3 | m | SC (80%) | chr2:47630386 | MSH2 | 59 | p.(Phe22fs) | 1835 | 1 | NM_000251.2 | c.62_63ins | Pathogenic (class 5) |
| 4 | m | SCS (80%) | chr2:47637361 | MSH2 | 42 | p.(Tyr165*) | 1999 | 3 | NM_000251.2 | c.495T>A | Pathogenic (class 5) |
| chr3:178952085 | PIK3CA | 36 | p.(His1047Arg) | 1658 | 21 | NM_006218.3 | c.3140A>G | Pathogenic (class 5) | |||
| chr9:139418362 | NOTCH1 | 25 | p.(Asn70fs) | 4684 | 3 | NM_017617.4 | c.209_210ins | Likely pathogenic (class 4) | |||
| chr9:139418364 | NOTCH1 | 40 | p.(Asn70fs) | 1996 | 3 | NM_017617.4 | c.207_208ins | Likely pathogenic (class 4) | |||
| chr10:89711899 | PTEN | 46 | p.(Arg173Cys) | 1797 | 6 | NM_000314.6 | c.517C>T | Likely pathogenic (class 4) | |||
| chr12:133202239 | POLE | 17 | p.(Gln2217*) | 128 | 47 | NM_006231.3 | c.6649C>T | Pathogenic (class 5) | |||
| chr17:7578403 | TP53 | 39 | p.(Cys176Tyr) | 1998 | 5 | NM_000546.5 | c.527G>A | Likely pathogenic (class 4) | |||
| chr17:29665125 | NF1 | 43 | p.(Gln2263*) | 2000 | 45 | NM_001042492.2 | c.6787C>T | Pathogenic (class 5) | |||
| 5 | f | SC (60%) | chr2:47693918 | MSH2 | 42 | p.(Gln545*) | 144 | 10 | NM_000251.2 | c.1632_1633del | Pathogenic (class 5) |
| 6 | m | SC (70%) | chr2:47693799 | MSH2 | 36 | p.(Pro507fs) | 2283 | 10 | NM_000251.2 | c.1518_1519ins | Pathogenic (class 5) |
| chr2:47693804 | MSH2 | 72 | p.(Pro507fs) | 1200 | 10 | NM_000251.2 | c.1518_1519ins | Pathogenic (class 5) | |||
| chr16:23637644 | PALB2 | 4 | p.(Ile887fs) | 637 | 7 | NM_024675.3 | c.2659_2660del | Uncertain significance (class 3) | |||
| 7 | m | SA (30%) | chr1:120466424 | NOTCH2 | 40 | p.(Arg1567fs) | 1944 | 26 | NM_024408.3 | c.4694_4695ins | Likely pathogenic (class 4) |
| chr2:47698188 | MSH2 | 82 | p.(Asn583fs) | 1984 | 11 | NM_000251.2 | c.1747_1748del | Pathogenic (class 5) | |||
| chr9:139402690 | NOTCH1 | 41 | p.(Arg1107*) | 901 | 20 | NM_017617.4 | c.3319C>T | Pathogenic (class 5) | |||
| chr11:534289 | HRAS | 43 | p.(Gly12Ser) | 1188 | 2 | NM_001130442.2 | c.34G>A | Uncertain significance (class 3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simic, D.; Dummer, R.; Freiberger, S.N.; Ramelyte, E.; Barysch, M.-J. Clinical and Molecular Features of Skin Malignancies in Muir-Torre Syndrome. Genes 2021, 12, 781. https://doi.org/10.3390/genes12050781
Simic D, Dummer R, Freiberger SN, Ramelyte E, Barysch M-J. Clinical and Molecular Features of Skin Malignancies in Muir-Torre Syndrome. Genes. 2021; 12(5):781. https://doi.org/10.3390/genes12050781
Chicago/Turabian StyleSimic, Dario, Reinhard Dummer, Sandra N. Freiberger, Egle Ramelyte, and Marjam-Jeanette Barysch. 2021. "Clinical and Molecular Features of Skin Malignancies in Muir-Torre Syndrome" Genes 12, no. 5: 781. https://doi.org/10.3390/genes12050781
APA StyleSimic, D., Dummer, R., Freiberger, S. N., Ramelyte, E., & Barysch, M.-J. (2021). Clinical and Molecular Features of Skin Malignancies in Muir-Torre Syndrome. Genes, 12(5), 781. https://doi.org/10.3390/genes12050781