
Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow with Clinical Decision Rules


Abstract
:1. Introduction
2. BRCA2 Protein, Functions, and Patient Mutations
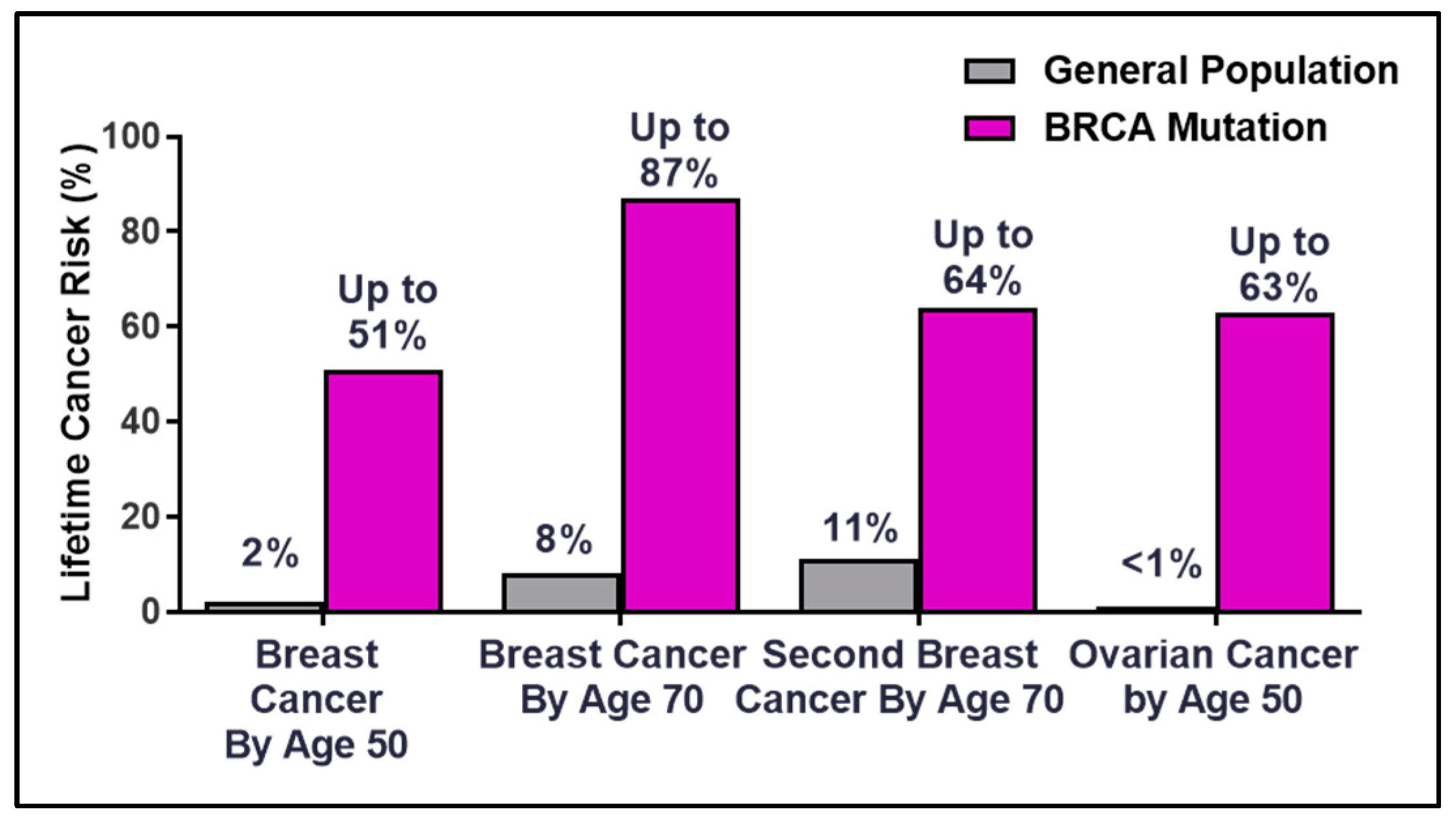
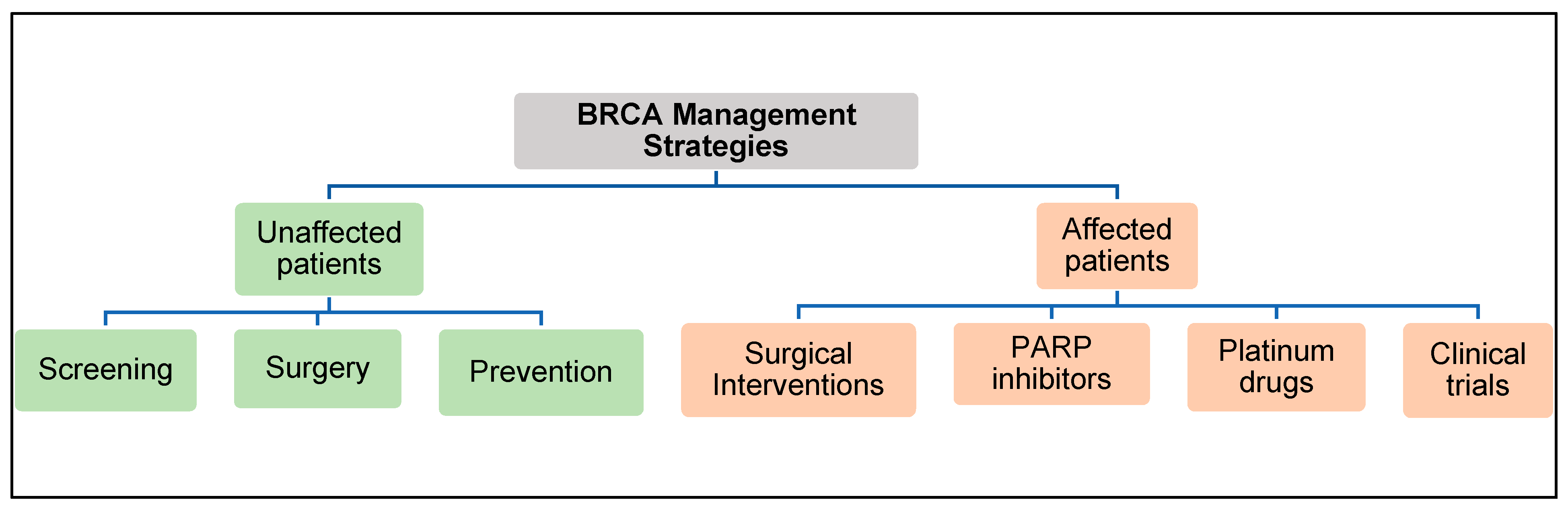
3. Clinical Management of Patients
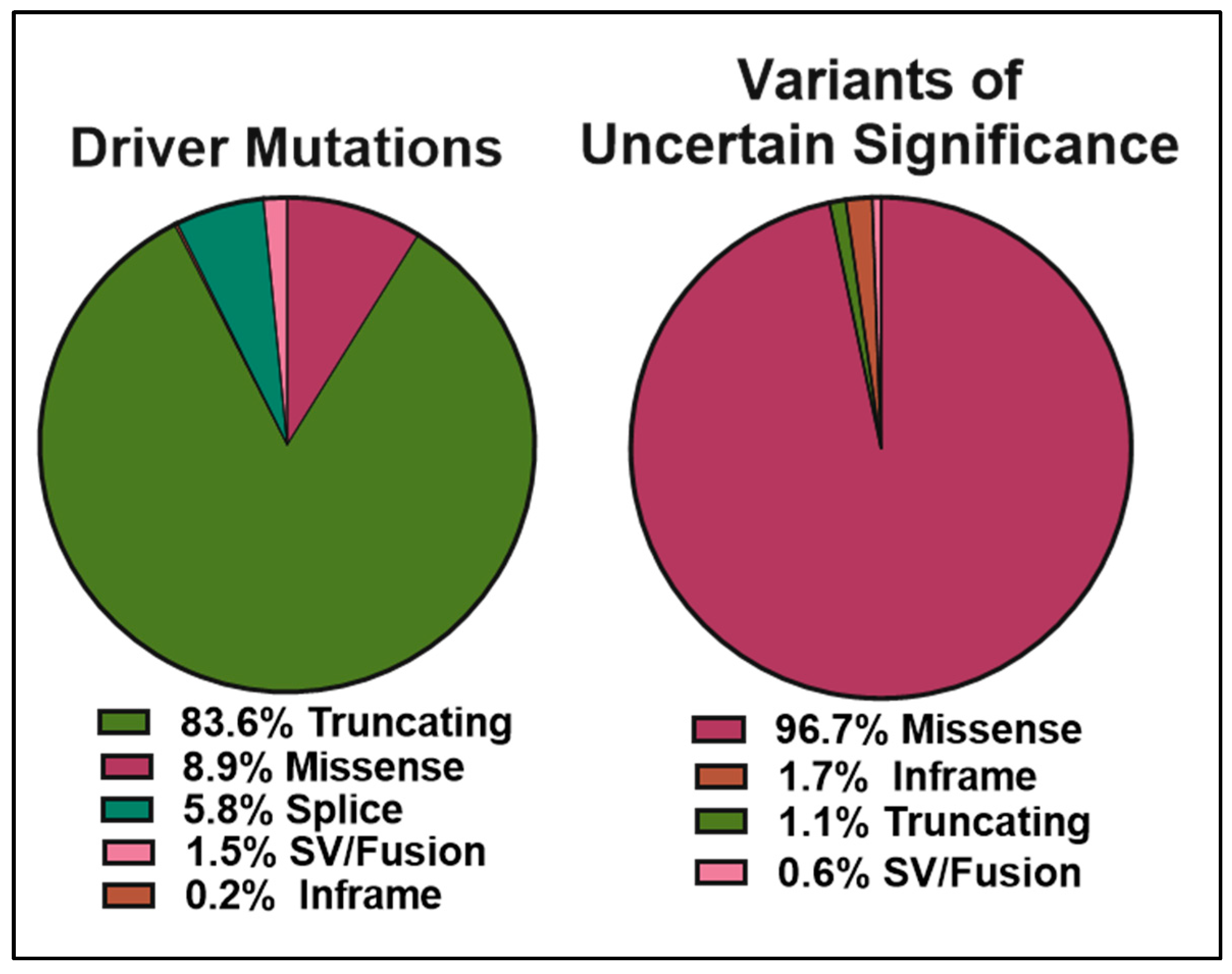
4. Curation of BRCA2 VUS
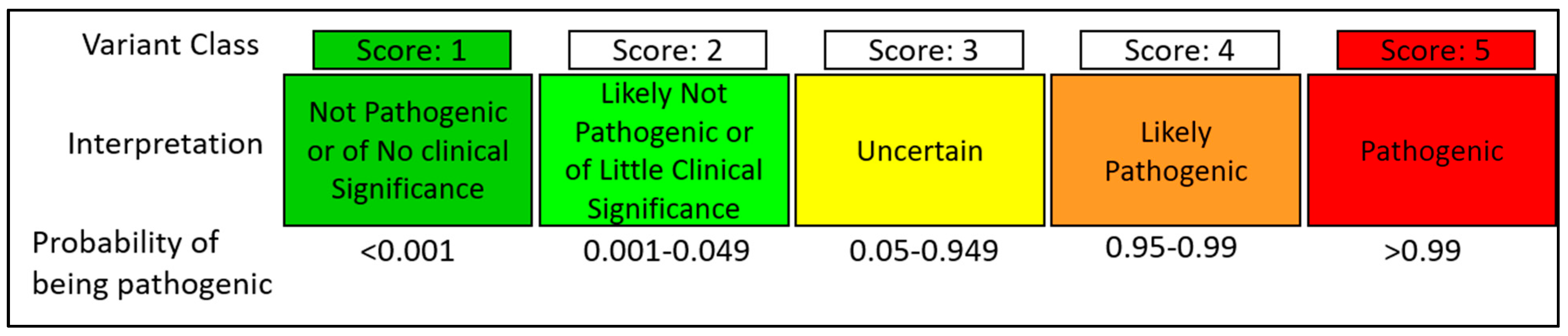
5. Methods of Classification
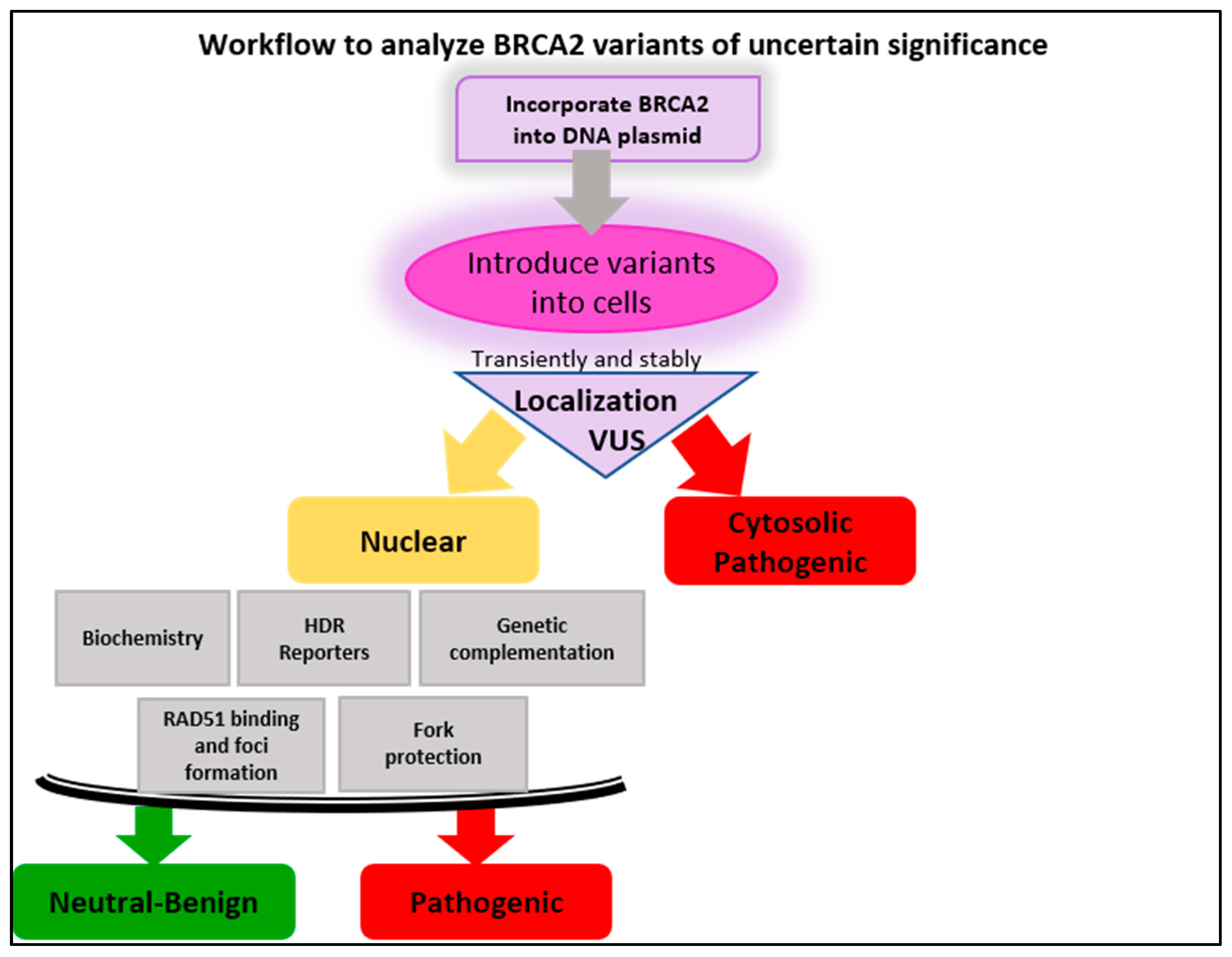
6. Functional Studies
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene brca2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.; Anderson, L.A.; Friedman, L.S.; Black, D.M.; Osborne-Lawrence, S.; Rowell, S.E.; Hall, J.M.; Solomon, E.; King, M.C. THRA1 and D17S183 flank an interval of <4 cm for the breast-ovarian cancer gene (BRCA1) on chromosome 17q21. Am. J. Hum. Genet. 1993, 52, 718–722. [Google Scholar]
- Nathanson, K.L.; Wooster, R.; Weber, B.L. Breast cancer genetics: What we know and what we need. Nat. Med. 2001, 7, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, N.; Goldgar, D.E.; Easton, D.F.; Rookus, M.; Brohet, R.; Antoniou, A.C.; Chang-Claude, J. Pregnancies, breast-feeding, and breast cancer risk in the international brca1/2 carrier cohort study (ibccs). J. Natl. Cancer Inst. 2006, 98, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, C.A.; Lubinski, J.; Neuhausen, S.L.; Ghadirian, P.; Lynch, H.T.; Isaacs, C.; Weber, B.; Moller, P.; Offit, K.; Kim-Sing, C.; et al. Effect of pregnancy as a risk factor for breast cancer inBRCA1/BRCA2 mutation carriers. Int. J. Cancer 2005, 117, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.; Lubinski, J.; Lynch, H.T.; Ghadirian, P.; Foulkes, W.D.; Kim-Sing, C.; Neuhausen, S.; Tung, N.; Rosen, B.; Gronwald, J.; et al. Family History of Cancer and Cancer Risks in Women with BRCA1 or BRCA2 Mutations. J. Natl. Cancer Inst. 2010, 102, 1874–1878. [Google Scholar] [CrossRef] [Green Version]
- Milne, R.L.; Osorio, A.; Cajal, T.R.Y.; Baiget, M.; Lasa, A.; Diaz-Rubio, E.; De La Hoya, M.; Caldés, T.; Teulé, A.; Lázaro, C.; et al. Parity and the risk of breast and ovarian cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. Treat. 2009, 119, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.; Easton, D. Variation in Cancer Risks, by Mutation Position, in BRCA2 Mutation Carriers. Am. J. Hum. Genet. 2001, 68, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results From Prospective Analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.; Mooij, T.M.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; Goldgar, D.E.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Steele, L.; Fields, P.; Ormiston, W.; Averill, D.; Daly, P.A.; McManus, R.; Neuhausen, S.L.; Ford, D.; Wooster, R.; et al. Cancer Risks in Two Large Breast Cancer Families Linked to BRCA2 on Chromosome 13q12-13. Am. J. Hum. Genet. 1997, 61, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nat. Cell Biol. 2000, 408, 429–432. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer Susceptibility and the Functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.A.; Masson, J.-Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Powell, S.N.; Willers, H.; Xia, F. Brca2 keeps rad51 in line. High-fidelity homologous recombination prevents breast and ovarian cancer? Mol. Cell 2002, 10, 1262–1263. [Google Scholar] [CrossRef]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The brca2 homologue brh2 nucleates rad51 filament formation at a dsdna-ssdna junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef]
- Yuan, S.S.; Lee, S.Y.; Chen, G.; Song, M.; Tomlinson, G.E.; Lee, E.Y. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999, 59, 3547–3551. [Google Scholar]
- Eckelmann, B.J.; Bacolla, A.; Wang, H.; Ye, Z.; Guerrero, E.N.; Jiang, W.; El-Zein, R.; Hegde, M.L.; Tomkinson, A.E.; Tainer, J.A.; et al. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2020, 2, zcaa013. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Lopes, M. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nat. Cell Biol. 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Shivji, M.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.J.; Wang, Y.; Lee, M.; Venkitaraman, A.R. Abnormal Cytokinesis in Cells Deficient in the Breast Cancer Susceptibility Protein BRCA2. Science 2004, 306, 876–879. [Google Scholar] [CrossRef]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 Localization to the Midbody by Filamin A Regulates CEP55 Signaling and Completion of Cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Hammel, M.; Yu, X.; Galkin, V.E.; Liu, D.; Tsai, M.-S.; et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010, 17, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.K.; Tischkowitz, M.; Simhadri, S.; Boshari, T.; Zayed, N.; Burke, K.A.; Berman, S.H.; Blecua, P.; Riaz, N.; Huo, Y.; et al. Compromised BRCA1–PALB2 interaction is associated with breast cancer risk. Oncogene 2017, 36, 4161–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.; Hsu, F.; Gilks, B.; et al. EMSY Links the BRCA2 Pathway to Sporadic Breast and Ovarian Cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.; Capanu, M.; Sabbaghian, N.; Li, L.; Liang, X.; Vallée, M.P.; Tavtigian, S.V.; Concannon, P.; Foulkes, W.D.; Bernstein, L.; et al. Rare germline mutations inPALB2and breast cancer risk: A population-based study. Hum. Mutat. 2012, 33, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of rad51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, G.; Jimenez-Sainz, J.; Presti, T.; Nguyen, T.; Jensen, R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016, 44, 5256–5270. [Google Scholar] [CrossRef] [Green Version]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef] [Green Version]
- Thorslund, T.; Esashi, F.; West, S.C. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J. 2007, 26, 2915–2922. [Google Scholar] [CrossRef] [Green Version]
- Marston, N.J.; Richards, W.J.; Hughes, D.; Bertwistle, D.; Marshall, C.J.; Ashworth, A. Interaction between the Product of the Breast Cancer Susceptibility Gene BRCA2 and DSS1, a Protein Functionally Conserved from Yeast to Mammals. Mol. Cell. Biol. 1999, 19, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Vaithiyalingam, S.; Filippo, J.S.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-ssDNA Structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-L.; Chen, C.-F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.-H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuta, R.; LaSalle, J.; Cheng, H.-L.; Shinohara, A.; Ogawa, H.; Copeland, N.; Jenkins, N.A.; Lalande, M.; Alt, F.W. RAB22 and RAB163/mouse BRCA2: Proteins that specifically interact with the RAD51 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6927–6932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nat. Cell Biol. 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nat. Cell Biol. 1997, 386, 804–810. [Google Scholar] [CrossRef]
- Wong, A.K.C.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 Interacts with the Evolutionarily Conserved BRC Motifs in the Human Breast Cancer Susceptibility Gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273. [Google Scholar] [CrossRef]
- Bertwistle, D.; Swift, S.; Marston, N.J.; Jackson, L.E.; Crossland, S.; Crompton, M.R.; Marshall, C.J.; Ashworth, A. Nuclear location and cell cycle regulation of the BRCA2 protein. Cancer Res. 1997, 57, 5485–5488. [Google Scholar] [PubMed]
- Han, X.; Saito, H.; Miki, Y.; Nakanishi, A. A CRM1-mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene 2007, 27, 2969–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spain, B.H.; Larson, C.J.; Shihabuddin, L.S.; Gage, F.H.; Verma, I.M. Truncated BRCA2 is cytoplasmic: Implications for cancer-linked mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 13920–13925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, K.-I.; Morotomi, K.; Saito, H.; Kato, M.; Matsuo, F.; Miki, Y. Nuclear Localization Signals of the BRCA2 Protein. Biochem. Biophys. Res. Commun. 2000, 270, 171–175. [Google Scholar] [CrossRef]
- Jeyasekharan, A.; Liu, Y.; Hattori, H.; Pisupati, V.; Jonsdottir, A.B.; Rajendra, E.; Lee, M.; Sundaramoorthy, E.; Schlachter, S.; Kaminski, C.F.; et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 2013, 20, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Silver, D.; Cantor, S.; Livingston, D.M.; Scully, R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999, 59, 1752–1756. [Google Scholar]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 Is Required for Homology-Directed Repair of Chromosomal Breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.J.; Yu, V.P.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.; Venkitaraman, A.R. Involvement of Brca2 in DNA Repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Tutt, A.; Gabriel, A.; Bertwistle, D.; Connor, F.; Paterson, H.; Peacock, J.; Ross, G.; Ashworth, A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol. 1999, 9, 1107–1110. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable Interaction between the Products of the BRCA1 and BRCA2 Tumor Suppressor Genes in Mitotic and Meiotic Cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Hucl, T.; Rago, C.; Gallmeier, E.; Brody, J.R.; Gorospe, M.; Kern, S.E. A Syngeneic Variance Library for Functional Annotation of Human Variation: Application to BRCA2. Cancer Res. 2008, 68, 5023–5030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional Restoration of BRCA2 Protein by Secondary BRCA2 Mutations in BRCA2-Mutated Ovarian Carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; De La Pompa, J.L.; Hakem, R.; Elia, A.; Yoshida, R.; Mo, R.; Nishina, H.; Chuang, T.; Wakeham, A.; Itie, A.; et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 1997, 11, 1242–1252. [Google Scholar] [CrossRef] [Green Version]
- Wiegant, W.W.; Overmeer, R.M.; Godthelp, B.C.; van Buul, P.P.; Zdzienicka, M.Z. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat. Res. Mol. Mech. Mutagen. 2006, 600, 79–88. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.; Neuhausen, S.L.; Hansen, T.V.; Couch, F.J.; Vreeswijk, M.P.; et al. Functional Assays for Analysis of Variants of Uncertain Significance inBRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karchin, R.; Agarwal, M.; Sali, A.; Couch, F.; Beattie, M.S. Classifying Variants of Undetermined Significance in BRCA2 with Protein Likelihood Ratios. Cancer Inform. 2008, 6, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Abul-Husn, N.S.; Team, C.G.; Soper, E.; Odgis, J.A.; Cullina, S.; Bobo, D.; Moscati, A.; Rodriguez, J.E.; Loos, R.; Cho, J.H.; et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019, 12, 2. [Google Scholar] [CrossRef]
- Neuhausen, S.L.; Gilewski, T.; Norton, L.; Tran, T.; McGuire, P.; Swensen, J.; Hampel, H.; Borgen, P.I.; Brown, K.L.; Skolnick, M.; et al. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat. Genet. 1996, 13, 126–128. [Google Scholar] [CrossRef]
- Sarantaus, L.; Huusko, P.; Eerola, H.; Launonen, V.; Vehmanen, P.; Rapakko, K.; Gillanders, E.M.; Syrjäkoski, K.; Kainu, T.; Vahteristo, P.; et al. Multiple founder effects and geographical clustering of BRCA1 and BRCA2 families in Finland. Eur. J. Hum. Genet. 2000, 8, 757–763. [Google Scholar] [CrossRef]
- Seong, M.-W.; Cho, S.; Noh, D.-Y.; Han, W.; Kim, S.-W.; Park, C.-M.; Park, H.-W.; Kim, J.Y.; Park, S.S. Comprehensive mutational analysis ofBRCA1/BRCA2for Korean breast cancer patients: Evidence of a founder mutation. Clin. Genet. 2009, 76, 152–160. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Trevino, V. Hotspotannotations-a database for hotspot mutations and annotations in cancer. Database (Oxford) 2020, 2020, baaa025. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location of Mutation in BRCA2 Gene and Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2018, 24, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dines, J.N.; Shirts, B.H.; Slavin, T.P.; Walsh, T.; King, M.-C.; Fowler, D.M.; Pritchard, C.C.; Shirts, B.H.; Pritchard, C.C. Systematic misclassification of missense variants in BRCA1 and BRCA2 “coldspots”. Genet. Med. 2020, 22, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruthi, S.; Gostout, B.S.; Lindor, N.M. Identification and management of women with brca mutations or hereditary predisposition for breast and ovarian cancer. Mayo Clin. Proc. 2010, 85, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhab, M.; Bismohun, S.; Mokbel, K. Risk-reducing strategies for women carrying brca1/2 mutations with a focus on prophylactic surgery. BMC Womens Health 2010, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.R.; Barwell, J.; Eccles, D.M.; Collins, A.; Izatt, L.; Jacobs, C.; Donaldson, A.; Brady, A.F.; Cuthbert, A.; Harrison, R.; et al. The Angelina Jolie effect: How high celebrity profile can have a major impact on provision of cancer related services. Breast Cancer Res. 2014, 16, 442. [Google Scholar] [CrossRef]
- James, P.A.; Mitchell, G.; Bogwitz, M.; Lindeman, G.J. The Angelina Jolie effect. Med. J. Aust. 2013, 199, 646. [Google Scholar] [CrossRef]
- Welsh, J.L.; Hoskin, T.L.; Day, C.N.; Thomas, A.S.; Cogswell, J.A.; Couch, F.J.; Boughey, J.C. Clinical Decision-Making in Patients with Variant of Uncertain Significance in BRCA1 or BRCA2 Genes. Ann. Surg. Oncol. 2017, 24, 3067–3072. [Google Scholar] [CrossRef]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in brca1 or brca2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.L.; Monteiro, A.N.A.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA-Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Goldgar, D.E.; Boffetta, P.; for the IARC Unclassified Genetic Variants Working Group. Assessing pathogenicity: Overview of results from the IARC Unclassified Genetic Variants Working Group. Hum. Mutat. 2008, 29, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.P.; Monteiro, A.N.A.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; Allen-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.; Iversen, E.S.; Couch, F.J.; et al. A Systematic Genetic Assessment of 1,433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer–Predisposition Genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, G.; Berry, D.A.; Aguilar, O. Determining Carrier Probabilities for Breast Cancer–Susceptibility Genes BRCA1 and BRCA2. Am. J. Hum. Genet. 1998, 62, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrer, J.; Duffy, S.W.; Cuzick, J. A breast cancer prediction model incorporating familial and personal risk factors. Stat. Med. 2004, 23, 1111–1130. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, A.C.; Cunningham, A.P.; Peto, J.; Evans, D.G.; Lalloo, F.; Narod, S.A.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Southey, M.C.; et al. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: Updates and extensions. Br. J. Cancer 2008, 98, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- García, E.B.G.; Oosterwijk, J.C.; Timmermans, M.; van Asperen, C.J.; Hogervorst, F.B.; Hoogerbrugge, N.; Vreeswijk, M.P. A method to assess the clinical significance of unclassified variants in the brca1 and brca2 genes based on cancer family history. Breast Cancer Res. 2009, 11, R8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurdle, A.B.; Lakhani, S.R.; Healey, S.; Parry, S.; Da Silva, L.M.; Brinkworth, R.; Hopper, J.L.; Brown, M.A.; Babikyan, D.; Chenevix-Trench, G.; et al. Clinical Classification of BRCA1 and BRCA2 DNA Sequence Variants: The Value of Cytokeratin Profiles and Evolutionary Analysis—A Report From the kConFab Investigators. J. Clin. Oncol. 2008, 26, 1657–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spearman, A.D.; Sweet, K.; Zhou, X.-P.; McLennan, J.; Couch, F.J.; Toland, A.E. Clinically Applicable Models to Characterize BRCA1 and BRCA2 Variants of Uncertain Significance. J. Clin. Oncol. 2008, 26, 5393–5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, L.; Vreeswijk, M.P.; Oldenburg, R.; Ouweland, A.V.D.; Oosterwijk, J.C.; Van Der Hout, A.H.; Hoogerbrugge, N.; Ligtenberg, M.; Ausems, M.G.; Van Der Luijt, R.B.; et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer 2009, 9, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- Eccles, D.M.; Mitchell, G.; Monteiro, A.N.A.; Schmutzler, R.; Couch, F.J.; Spurdle, A.B.; Gómez-García, E.B.; Driessen, R.; Lindor, N.; Blok, M.; et al. BRCA1 and BRCA2 genetic testing—pitfalls and recommendations for managing variants of uncertain clinical significance. Ann. Oncol. 2015, 26, 2057–2065. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Byrnes, G.B.; Goldgar, D.E.; Thomas, A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 2008, 29, 1342–1354. [Google Scholar] [CrossRef] [Green Version]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S.; for the IARC Unclassified Genetic Variants Working Group. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Biswas, K.; Das, R.; Alter, B.P.; Kuznetsov, S.G.; Stauffer, S.; North, S.L.; Burkett, S.; Brody, L.C.; Meyer, S.; Byrd, R.A.; et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell–based assay. Blood 2011, 118, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Das, R.; Eggington, J.M.; Qiao, H.; North, S.L.; Stauffer, S.; Burkett, S.; Martin, B.K.; Southon, E.; Sizemore, S.C.; et al. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum. Mol. Genet. 2012, 21, 3993–4006. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Lipton, G.B.; Stauffer, S.; Sullivan, T.; Cleveland, L.; Southon, E.; Reid, S.; Magidson, V.; Edwin, S.I., Jr.; Sharan, S.K. A computational model for classification of BRCA2 variants using mouse embryonic stem cell-based functional assays. NPJ Genom. Med. 2020, 5, 52. [Google Scholar] [CrossRef]
- Carvalho, M.; Couch, F.J.; Monteiro, A.N. Functional assays for BRCA1 and BRCA2. Int. J. Biochem. Cell Biol. 2007, 39, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Rasmussen, L.J.; Hofstra, R.; Monteiro, A.N.; Greenblatt, M.S.; de Wind, N. Iarc Unclassified Genetic for the IARC Unclassified Genetic Variants Working Group Assessment of functional effects of unclassified genetic variants. Hum. Mutat. 2008, 29, 1314–1326. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, D.J.; Agarwal, M.K.; Pankratz, V.S.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wadum, L.; Johnson, K.; Mentlick, J.; Tavtigian, S.V.; et al. Functional Assays for Classification of BRCA2 Variants of Uncertain Significance. Cancer Res. 2008, 68, 3523–3531. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, S.G.; Liu, P.; Sharan, S.K. Mouse embryonic stem cell–based functional assay to evaluate mutations in BRCA2. Nat. Med. 2008, 14, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Guidugli, L.; Pankratz, V.S.; Singh, N.; Thompson, J.; Erding, C.A.; Engel, C.; Schmutzler, R.; Domchek, S.; Nathanson, K.; Radice, P.; et al. A Classification Model for BRCA2 DNA Binding Domain Missense Variants Based on Homology-Directed Repair Activity. Cancer Res. 2012, 73, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidugli, L.; Shimelis, H.; Masica, D.L.; Pankratz, V.S.; Lipton, G.B.; Singh, N.; Hu, C.; Monteiro, A.N.A.; Lindor, N.M.; Goldgar, D.E.; et al. Assessment of the clinical relevance of brca2 missense variants by functional and computational approaches. Am. J. Hum. Genet 2018, 102, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, G.; Morolli, B.; Calléja, F.M.; Plomp, A.; Mesman, R.L.; Meijers, M.; Sharan, S.K.; Vreeswijk, M.P.; Vrieling, H. An efficient pipeline for the generation and functional analysis of human BRCA2 variants of uncertain significance. Hum. Mutat. 2014, 35, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Mesman, R.; Msc, F.M.G.R.C.; Hendriks, G.; Morolli, B.; Misovic, B.; Devilee, P.; Van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. The functional impact of variants of uncertain significance in BRCA2. Genet. Med. 2019, 21, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, M.E.; Hu, C.; Lee, K.Y.; LaDuca, H.; Fulk, K.; Durda, K.M.; Deckman, A.M.; Goldgar, D.E.; Monteiro, A.N.; Gnanaolivu, R.; et al. Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am. J. Hum. Genet. 2021, 108, 458–468. [Google Scholar] [CrossRef]
- Shimelis, H.; Mesman, R.; Von Nicolai, C.; Ehlen, A.; Guidugli, L.; Martin, C.; Calléja, F.M.; Meeks, H.; Hallberg, E.; Hinton, J.; et al. BRCA2 Hypomorphic Missense Variants Confer Moderate Risks of Breast Cancer. Cancer Res. 2017, 77, 2789–2799. [Google Scholar] [CrossRef] [Green Version]
- Balìa, C.; Galli, A.; Caligo, M.A. Effect of the overexpression of BRCA2 unclassified missense variants on spontaneous homologous recombination in human cells. Breast Cancer Res. Treat. 2011, 129, 1001–1009. [Google Scholar] [CrossRef]
- Nakanishi, A.; Han, X.; Saito, H.; Taguchi, K.; Ohta, Y.; Imajoh-Ohmi, S.; Miki, Y. Interference with BRCA2, which localizes to the centrosome during S and early M phase, leads to abnormal nuclear division. Biochem. Biophys. Res. Commun. 2007, 355, 34–40. [Google Scholar] [CrossRef]
- Spugnesi, L.; Balia, C.; Collavoli, A.; Falaschi, E.; Quercioli, V.; Caligo, M.A.; Galli, A. Effect of the expression of BRCA2 on spontaneous homologous recombination and DNA damage-induced nuclear foci in Saccharomyces cerevisiae. Mutagenesis 2013, 28, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Thomassen, M.; Blanco, A.; Montagna, M.; Hansen, T.V.O.; Pedersen, I.S.; Gutiérrez-Enríquez, S.; Menéndez, M.; Fachal, L.; Santamariña, M.; Steffensen, A.Y.; et al. Characterization of BRCA1 and BRCA2 splicing variants: A collaborative report by ENIGMA consortium members. Breast Cancer Res. Treat. 2011, 132, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar] [PubMed]
- Fraile-Bethencourt, E.; Díez-Gómez, B.; Velásquez-Zapata, V.; Acedo, A.; Sanz, D.J.; Velasco, E.A. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017, 13, e1006691. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Acedo, A.; Velasco, E.A. Identification of Eight Spliceogenic Variants in BRCA2 Exon 16 by Minigene Assays. Front. Genet. 2018, 9, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, D.J.; Acedo, A.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Esteban-Cardeñosa, E.; Lastra, E.; Pagani, F.; Miner, C.; Velasco, E.A. A High Proportion of DNA Variants of BRCA1 and BRCA2 Is Associated with Aberrant Splicing in Breast/Ovarian Cancer Patients. Clin. Cancer Res. 2010, 16, 1957–1967. [Google Scholar] [CrossRef] [Green Version]
- Wangensteen, T.; Felde, C.N.; Ahmed, D.; Mæhle, L.; Ariansen, S.L. Diagnostic mRNA splicing assay for variants in BRCA1 and BRCA2 identified two novel pathogenic splicing aberrations. Hered. Cancer Clin. Pr. 2019, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, M.; Kohsaka, S.; Ueno, T.; Momozawa, Y.; Inoue, S.; Tamura, K.; Shimomura, A.; Hosoya, N.; Kobayashi, H.; Tanaka, S.; et al. High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat. Commun. 2020, 11, 2573. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nat. Cell Biol. 2018, 562, 217–222. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study/Consortium | Description |
|---|---|
| CIMBA | Consortium of investigators of modifiers of BRCA1/2 was established in 2005 |
| PROSE | Consortium in the prevention and observation of surgical endpoints [81] |
| ENIGMA | International consortium of investigators to determine the clinical significance of sequence variants in BRCA1, BRCA2 and other known or suspected breast cancer genes (www.enigmaconsortium.org, accessed on 15 April 2021) [82] |
| EMBRACE | Study aims to create a register of BRCA1 and BRCA2 families with defects in other genes to find out the cancer risk in these associations [10] |
| IBCCS | International BRCA1/2 carrier cohort study: purpose, rationale, and study design (https://breast-cancer-research.biomedcentral.com/articles/10.1186/bcr93, accessed on 15 April 2021) |
| Breast Cancer Linkage Consortium | International data sharing platform established in 1989 to collect families with breast cancer by linkage analysis |
| PROMPT | Prospective Registry of Multiplex Testing for patients and their families (www.promptstudy.org, accessed on 15 April 2021) |
| BRCA2 Databases | Total Variants | Variants of Uncertain Significance | Missense Variants | Pathogenic Variants |
|---|---|---|---|---|
| Breast Cancer Information Core (BIC) (https://research.nhgri.nih.gov/bic/, accessed on 15 April 2021) | 14,914 |  | 7156 (48%) | |
| Leiden Open Variant Database (LOVD) (https://www.lovd.nl, accessed on 15 April 2021) | 920 | | 871 (94.6%) | |
| Catalogue of Somatic Mutation in Cancer (COSMIC) (https://cancer.sanger.ac.uk/cosmic, accessed on 15 April 2021) | 2729 | | 1548 (56.72%) | |
| BRCA Exchange (https://brcaexchange.org, accessed on 15 April 2021) | 20,751 | 16,649 (83%) | | 2672 (13%) |
| ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 15 April 2021) | 13,109 | 5381 (41%) | 5756 (44%) | 3780 (29%) |
Data unavailable.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez-Sainz, J.; Jensen, R.B. Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow with Clinical Decision Rules. Genes 2021, 12, 780. https://doi.org/10.3390/genes12050780
Jimenez-Sainz J, Jensen RB. Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow with Clinical Decision Rules. Genes. 2021; 12(5):780. https://doi.org/10.3390/genes12050780
Chicago/Turabian StyleJimenez-Sainz, Judit, and Ryan B. Jensen. 2021. "Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow with Clinical Decision Rules" Genes 12, no. 5: 780. https://doi.org/10.3390/genes12050780
APA StyleJimenez-Sainz, J., & Jensen, R. B. (2021). Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow with Clinical Decision Rules. Genes, 12(5), 780. https://doi.org/10.3390/genes12050780