
Identification of a Splice Variant (c.5074+3A>C) of BRCA1 by RNA Sequencing and TOPO Cloning

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Sequencing
2.2. Bioinformatics
2.3. RNA Analysis
2.4. TOPO Cloning and Sequencing of the BRCA1 c.5074+3A>C Variant
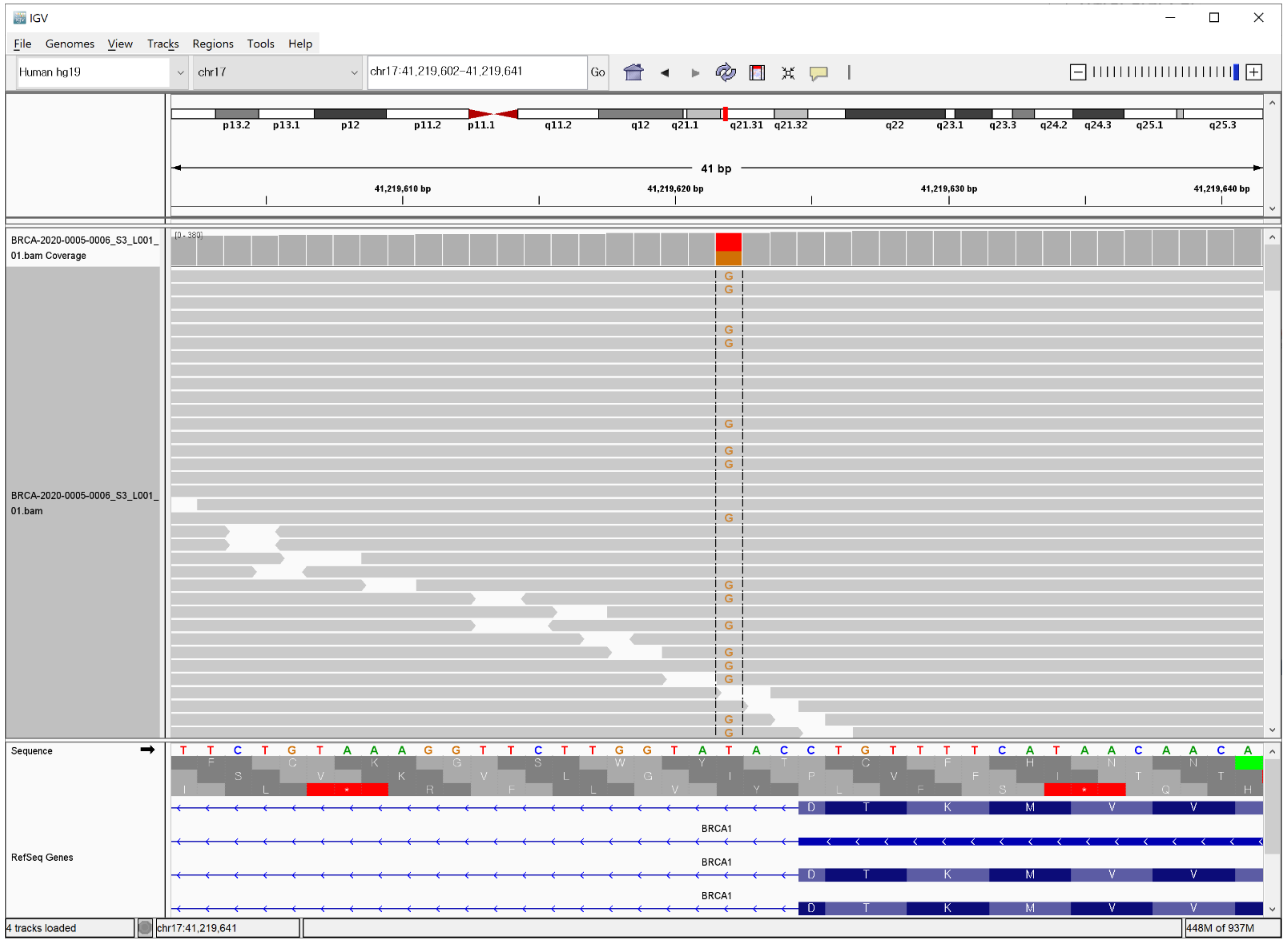
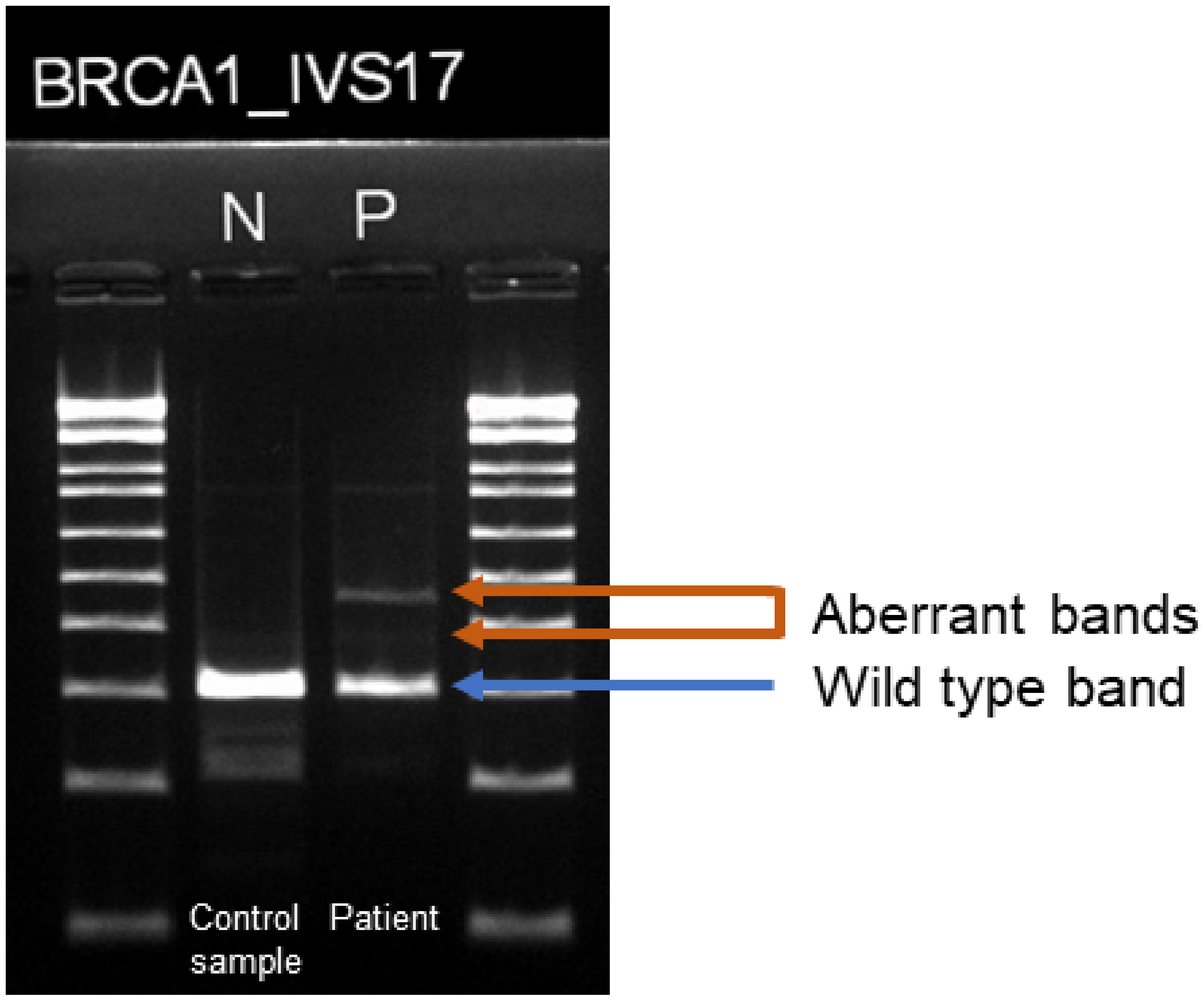
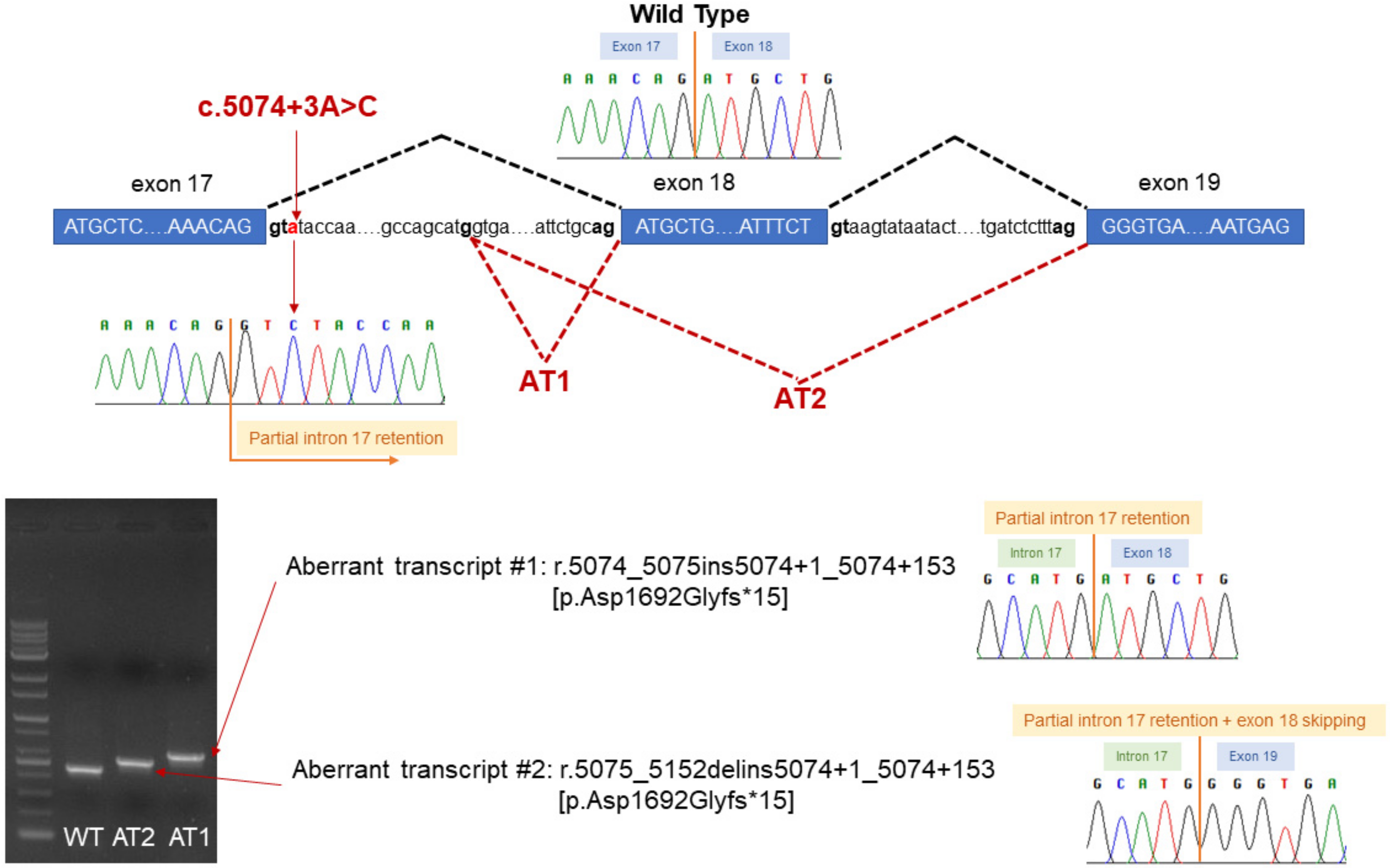
3. Case Description
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088. [Google Scholar] [CrossRef]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Gordon, O.K. Positive Results: Making the Best Decisions When You’re at High Risk for Breast or Ovarian Cancer; Prometheus Books: Amherst, NY, USA, 2010. [Google Scholar]
- Forbes, C.; Fayter, D.; de Kock, S.; Quek, R.G. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manag. Res. 2019, 11, 2321–2337. [Google Scholar] [CrossRef] [Green Version]
- González-Santiago, S.; Ramón y Cajal, T.; Aguirre, E.; Alés-Martínez, J.E.; Andrés, R.; Balmaña, J.; Graña, B.; Herrero, A.; Llort, G.; González-del-Alba, A.; et al. SEOM clinical guidelines in hereditary breast and ovarian cancer (2019). Clin. Transl. Oncol. 2020, 22, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.J.; Bressac-De Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Béroud, G.; et al. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum Mutat. 2016, 37, 1318–1328. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Culp, M.; Johnson, K.; Michailides, G. ada: An R Package for Stochastic Boosting. J. Stat. Softw. 2006, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and Regression by RandomForest. R News 2001, 23, 18–22. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, M.; Castellsagué, J.; Mirete, M.; Pros, E.; Feliubadaló, L.; Osorio, A.; Calaf, M.; Tornero, E.; del Valle, J.; Fernández-Rodríguez, J.; et al. Assessing the RNA effect of 26 DNA variants in the BRCA1 and BRCA2 genes. Breast Cancer Res. Treat. 2012, 132, 979–992. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [Green Version]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautz, D.; Renz, M. An optimized freeze-squeeze method for the recovery of DNA fragments from agarose gels. Anal. Biochem. 1983, 132, 14–19. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Roest, P.A.M.; Roberts, R.G.; Sugino, S.; van Ommen, G.-J.B.; den Dunnen, J.T. Protein truncation test (PTT) for rapid detection of translation-terminating mutations. Hum. Mol. Genet. 1993, 2, 1719–1721. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Neuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G.; et al. Functional Assays for Analysis of Variants of Uncertain Significance in BRCA2. Hum Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin-Vidoz, L.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Lenoir, G.M.; Mazoyer, S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002, 11, 2805–2814. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.; Kim, J.H.; Ahn, S.H.; Gu, H.; Chang, S.; Lee, W.; Kim, D.-Y.; Chun, S.; Min, W.-K. Identification of a Splice Variant (c.5074+3A>C) of BRCA1 by RNA Sequencing and TOPO Cloning. Genes 2021, 12, 810. https://doi.org/10.3390/genes12060810
Hong J, Kim JH, Ahn SH, Gu H, Chang S, Lee W, Kim D-Y, Chun S, Min W-K. Identification of a Splice Variant (c.5074+3A>C) of BRCA1 by RNA Sequencing and TOPO Cloning. Genes. 2021; 12(6):810. https://doi.org/10.3390/genes12060810
Chicago/Turabian StyleHong, Jinyoung, Ji Hyun Kim, Se Hee Ahn, Hyunjung Gu, Suhwan Chang, Woochang Lee, Dae-Yeon Kim, Sail Chun, and Won-Ki Min. 2021. "Identification of a Splice Variant (c.5074+3A>C) of BRCA1 by RNA Sequencing and TOPO Cloning" Genes 12, no. 6: 810. https://doi.org/10.3390/genes12060810
APA StyleHong, J., Kim, J. H., Ahn, S. H., Gu, H., Chang, S., Lee, W., Kim, D. -Y., Chun, S., & Min, W. -K. (2021). Identification of a Splice Variant (c.5074+3A>C) of BRCA1 by RNA Sequencing and TOPO Cloning. Genes, 12(6), 810. https://doi.org/10.3390/genes12060810