
Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses


Abstract
:1. Introduction
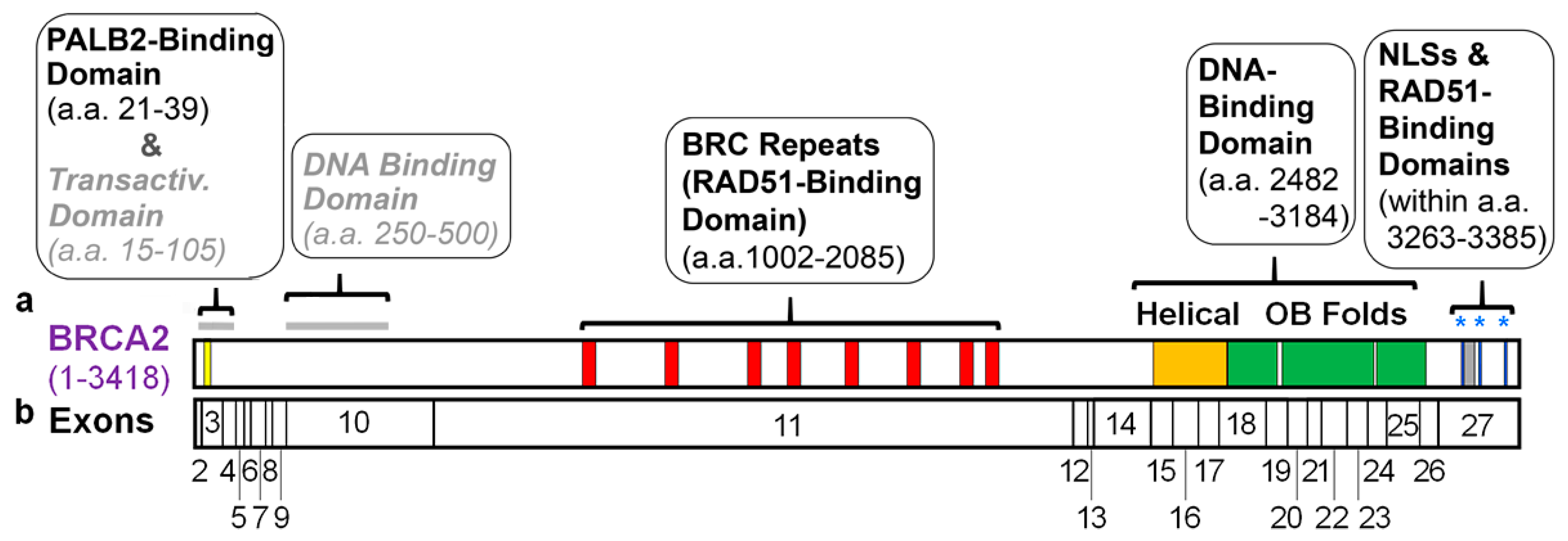
2. Known Domains within BRCA2
3. Understanding the Roles of Different Domains within BRCA2, and an Overview of Models/Systems Developed/Utilized to Elucidate These Functions
3.1. The Importance of Defining the Roles of Different Domains of BRCA2 for Understanding BRCA2 Function as a Tumor Suppressor and Harnessing Genetic Screens to Benefit Patients
3.2. Systems to Test BRCA2 Function Based upon the Expression of Mutants and Fragments of BRCA2, and Utilizing RNA Interference
3.2.1. Systems Based upon Gene Knockouts, RNAi, and the Use of Protein Fragments
3.2.2. Systems Based upon Ectopic Expression of Mutants of Full-Length BRCA2
4. The Function of Different Domains of BRCA2 in DNA Damage Responses
4.1. Domain-by-Domain Look at Roles of BRCA2 in DNA Damage Responses
4.2. Functional Relationships between Domains of BRCA2
5. Conclusions and an Outline of Parameters Necessary in an Ideal Expression System to Test the Function of Different Domains and Variants of BRCA2
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Cavallo, F.; Perrouault, L.; Giovannangeli, C.; Moynahan, M.E.; Barchi, M.; Brunet, E.; Jasin, M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat. Struct. Mol. Biol. 2011, 18, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Godthelp, B.C.; Wiegant, W.W.; Waisfisz, Q.; Medhurst, A.L.; Arwert, F.; Joenje, H.; Zdzienicka, M.Z. Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutat. Res. Mol. Mech. Mutagen. 2006, 594, 39–48. [Google Scholar] [CrossRef]
- Yuan, S.S.; Lee, S.Y.; Chen, G.; Song, M.; Tomlinson, G.E.; Lee, E.Y. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999, 59, 3547–3551. [Google Scholar] [PubMed]
- Abbott, D.; Holt, J.T.; Freeman, M. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J. Natl. Cancer Inst. 1998, 90, 978–985. [Google Scholar] [CrossRef]
- Al Abo, M.; Dejsuphong, D.; Hirota, K.; Yonetani, Y.; Yamazoe, M.; Kurumizaka, H.; Takeda, S. Compensatory functions and interdependency of the DNA-binding domain of BRCA2 with the BRCA1–PALB2–BRCA2 Complex. Cancer Res. 2014, 74, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.D.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; de Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Simhadri, S.; Vincelli, G.; Huo, Y.; Misenko, S.; Foo, T.K.; Ahlskog, J.; Sørensen, C.S.; Oakley, G.G.; Ganesan, S.; Bunting, S.F.; et al. PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response. Oncogene 2019, 38, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.; Nähse-Kumpf, V.; Kousholt, A.N.; Klein, D.K.; Lund-Andersen, C.; Lees, M.; Johansen, J.V.; Syljuåsen, R.G.; Sørensen, C.S. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep. 2011, 12, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, A.; O’Hara, C.; Khan, S.; Shack, L.; Woodward, E.; Maher, E.R.; Lalloo, F.; Evans, D.G.R. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam. Cancer 2012, 11, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; Lapolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; Goldgar, D.E.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- Lee, H. Cycling with BRCA2 from DNA repair to mitosis. Exp. Cell Res. 2014, 329, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Abidin, S.N.-Z.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2018, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Gacem, R.; Hachana, M.; Ziadi, S.; Amara, K.; Ksia, F.; Mokni, M.; Trimeche, M. Contribution of epigenetic alteration of BRCA1 and BRCA2 genes in breast carcinomas in Tunisian patients. Cancer Epidemiol. 2012, 36, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-N.; Tseng, R.-C.; Hsu, H.-S.; Chen, J.-Y.; Tzao, C.; Ho, W.L.; Wang, Y.-C. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non–small cell lung cancer. Clin. Cancer Res. 2007, 13, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Pal, R.; Srivastava, N.; Chopra, R.; Gochhait, S.; Gupta, P.; Prakash, N.; Agarwal, G.; Bamezai, R.N. Investigation of DNA damage response and apoptotic gene methylation pattern in sporadic breast tumors using high throughput quantitative DNA methylation analysis technology. Mol. Cancer 2010, 9, 303. [Google Scholar] [CrossRef] [Green Version]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Savage, S.A.; Walsh, M.F. Myelodysplastic syndrome, acute myeloid leukemia, and cancer surveillance in Fanconi anemia. Hematol. Clin. N. Am. 2018, 32, 657–668. [Google Scholar] [CrossRef]
- Wagner, J.E.; Tolar, J.; Levran, O.; Scholl, T.; Deffenbaugh, A.; Satagopan, J.; Ben-Porat, L.; Mah, K.; Batish, S.D.; Kutler, D.I.; et al. Germline mutations in BRCA2: Shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood 2004, 103, 3226–3229. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, B.; Shimamura, A.; Moreau, L.; Baldinger, S.; Hag-Alshiekh, M.; Bostrom, B.; Sencer, S.; D’Andrea, A.D. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004, 103, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Offit, K.; Levran, O.; Mullaney, B.; Mah, K.; Nafa, K.; Batish, S.D.; Diotti, R.; Schneider, H.; Deffenbaugh, A.; Scholl, T.; et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J. Natl. Cancer Inst. 2003, 95, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.; Brough, R.; Lord, C.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nat. Cell Biol. 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Mesman, R.L.S.; Calleja, F.; de la Hoya, M.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. Alternative mRNA splicing can attenuate the pathogenicity of presumed loss-of-function variants in BRCA2. Genet. Med. 2020, 22, 1355–1365. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nat. Cell Biol. 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Toland, A.E.; Andreassen, P.R. DNA repair-related functional assays for the classification of BRCA1 and BRCA2 variants: A critical review and needs assessment. J. Med. Genet. 2017, 54, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.W.; Swift, S.; Lord, C.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef] [Green Version]
- Rajendra, E.; Venkitaraman, A.R. Two modules in the BRC repeats of BRCA2 mediate structural and functional interactions with the RAD51 recombinase. Nucleic Acids Res. 2009, 38, 82–96. [Google Scholar] [CrossRef]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-ssDNA Structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- Davies, O.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51–DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar] [CrossRef]
- Spain, B.H.; Larson, C.J.; Shihabuddin, L.S.; Gage, F.H.; Verma, I.M. Truncated BRCA2 is cytoplasmic: Implications for cancer-linked mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 13920–13925. [Google Scholar] [CrossRef] [Green Version]
- von Nicolai, C.; Ehlén, Å.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.; Ponder, B.A.J.; Hughes-Davies, L.; Seltmann, M.; Kouzarides, T. Transcriptional activation functions in BRCA2. Nat. Cell Biol. 1997, 386, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Park, J.-Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [Green Version]
- Bignell, G.; Micklem, G.; Stratton, M.R.; Ashworth, A.; Wooster, R. The BRC repeats are conserved in mammalian BRCA2 proteins. Hum. Mol. Genet. 1997, 6, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Chen, C.-F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.-H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.K.C.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 Interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef] [Green Version]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, I.D.; Wadsworth, R.I.; Cubeddu, L.; Blankenfeldt, W.; Naismith, J.H.; White, M. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J. 2003, 22, 2561–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzin, A.G. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef]
- Marston, N.J.; Richards, W.J.; Hughes, D.; Bertwistle, D.; Marshall, C.J.; Ashworth, A. Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals. Mol. Cell. Biol. 1999, 19, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nat. Cell Biol. 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.; West, S. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.-I.; Morotomi, K.; Saito, H.; Kato, M.; Matsuo, F.; Miki, Y. Nuclear localization signals of the BRCA2 protein. Biochem. Biophys. Res. Commun. 2000, 270, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Das, R.; Alter, B.P.; Kuznetsov, S.; Stauffer, S.; North, S.L.; Burkett, S.; Brody, L.C.; Meyer, S.; Byrd, R.A.; et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 2011, 118, 2430–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirthagiri, E.; Klarmann, K.D.; Shukla, A.K.; Southon, E.; Biswas, K.; Martin, B.K.; North, S.L.; Magidson, V.; Burkett, S.; Haines, D.C.; et al. BRCA2 minor transcript lacking exons 4-7 supports viability in mice and may account for survival of humans with a pathogenic biallelic mutation. Hum. Mol. Genet. 2016, 25, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Moyer, V.A.; U.S. Preventive Services Task Force. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2014, 160, 271–281. [Google Scholar] [CrossRef]
- Willoughby, A.; Andreassen, P.R.; Toland, A.E. Genetic testing to guide risk-stratified screens for breast cancer. J. Pers. Med. 2019, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.; Ehlen, A.; Galli, A.; Monteiro, A.N.; Neuhausen, S.L.; Hansen, T.V.; Couch, F.J.; Vreeswijk, M.P.; et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Suh, D.H.; Lee, K.-H.; Kim, K.; Kang, S.; Kim, J.-W. Major clinical research advances in gynecologic cancer in 2014. J. Gynecol. Oncol. 2015, 26, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Raedler, L.A. Rubraca (Rucaparib) second parp inhibitor approved for patients with advanced, BRCA-positive ovarian cancer. Cancer Discov. 2017, 7, 120–121. [Google Scholar]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nat. Cell Biol. 1997, 386, 804–810. [Google Scholar] [CrossRef]
- Kuznetsov, S.; Liu, P.; Sharan, S.K. Mouse embryonic stem cell–based functional assay to evaluate mutations in BRCA2. Nat. Med. 2008, 14, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.M.; Jonkers, J. Mouse models of BRCA1 and BRCA2 deficiency: Past lessons, current understanding and future prospects. Oncogene 2006, 25, 5885–5897. [Google Scholar] [CrossRef] [Green Version]
- Bruun, N.; Folias, A.; Akkari, Y.; Cox, Y.; Olson, S.; Moses, R. siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair 2003, 2, 1007–1013. [Google Scholar] [CrossRef]
- Ohashi, A.; Zdzienicka, M.Z.; Chen, J.; Couch, F.J. Fanconi anemia complementation group D2 (FANCD2) Functions independently of BRCA2- and RAD51-associated homologous recombination in response to DNA damage. J. Biol. Chem. 2005, 280, 14877–14883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-F.; Chen, P.-L.; Zhong, Q.; Sharp, Z.D.; Lee, W.-H. Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G2/M checkpoint control. J. Biol. Chem. 1999, 274, 32931–32935. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Taghian, D.G.; DeFrank, J.S.; Zeng, Z.-C.; Willers, H.; Iliakis, G.; Powell, S.N. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc. Natl. Acad. Sci. USA 2001, 98, 8644–8649. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nat. Cell Biol. 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Andreassen, P.R.; D’Andrea, A.D. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, L.Y.; Ouchi, T.; Aaronson, S.A. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl. Acad. Sci. USA 1998, 95, 13869–13874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siaud, N.; Barbera, M.A.; Egashira, A.; Lam, I.; Christ, N.; Schlacher, K.; Xia, B.; Jasin, M. Plasticity of BRCA2 function in homologous recombination: Genetic interactions of the PALB2 and DNA binding domains. PLoS Genet. 2011, 7, e1002409. [Google Scholar] [CrossRef] [PubMed]
- Filippo, J.S.; Chi, P.; Sehorn, M.G.; Etchin, J.; Krejci, L.; Sung, P. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J. Biol. Chem. 2006, 281, 11649–11657. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, G.; Jimenez-Sainz, J.; Presti, T.; Nguyen, T.; Jensen, R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016, 44, 5256–5270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Vaithiyalingam, S.; Filippo, J.S.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, R.; Dion-Côté, A.-M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.-Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar] [PubMed]
- Richardson, M.E.; Hu, C.; Lee, K.Y.; LaDuca, H.; Fulk, K.; Durda, K.M.; Deckman, A.M.; Goldgar, D.E.; Monteiro, A.N.; Gnanaolivu, R.; et al. Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am. J. Hum. Genet. 2021, 108, 458–468. [Google Scholar] [CrossRef]
- Guidugli, L.; Shimelis, H.; Masica, D.L.; Pankratz, V.S.; Lipton, G.B.; Singh, N.; Hu, C.; Monteiro, A.N.A.; Lindor, N.M.; Goldgar, D.E.; et al. Assessment of the clinical relevance of BRCA2 missense variants by functional and computational approaches. Am. J. Hum. Genet. 2018, 102, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidugli, L.; Pankratz, V.S.; Singh, N.; Thompson, J.; Erding, C.A.; Engel, C.; Schmutzler, R.; Domchek, S.; Nathanson, K.; Radice, P.; et al. A Classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2012, 73, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Connor, F.; Smith, A.; Wooster, R.; Stratton, M.; Dixon, A.; Campbell, E.; Tait, T.-M.; Freeman, T.; Ashworth, A. Cloning, chromosomal mapping and expression pattern of the mouse Brca2 Gene. Hum. Mol. Genet. 1997, 6, 291–300. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadalo, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, K.; Das, R.; Eggington, J.M.; Qiao, H.; North, S.L.; Stauffer, S.; Burkett, S.S.; Martin, B.K.; Southon, E.; Sizemore, S.C.; et al. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum. Mol. Genet. 2012, 21, 3993–4006. [Google Scholar] [CrossRef] [Green Version]
- Mesman, R.L.S.; Calléja, F.M.G.R.; Hendriks, G.; Morolli, B.; Misovic, B.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. The functional impact of variants of uncertain significance in BRCA2. Genet. Med. 2019, 21, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Kohsaka, S.; Ueno, T.; Momozawa, Y.; Inoue, S.; Tamura, K.; Shimomura, A.; Hosoya, N.; Kobayashi, H.; Tanaka, S.; et al. High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Hucl, T.; Rago, C.; Gallmeier, E.; Brody, J.R.; Gorospe, M.; Kern, S.E. A syngeneic variance library for functional annotation of human variation: Application to BRCA2. Cancer Res. 2008, 68, 5023–5030. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150496. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nat. Cell Biol. 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Hartford, S.; Chittela, R.; Ding, X.; Vyas, A.; Martin, B.; Burkett, S.; Haines, D.C.; Southon, E.; Tessarollo, L.; Sharan, S.K. Interaction with PALB2 is essential for maintenance of genomic integrity by BRCA2. PLoS Genet. 2016, 12, e1006236. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.; Monteiro, A.N.A.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Approach | Purpose Developed for | Key Limitations | Refs.* |
|---|---|---|---|
| Expression of mini-BRCA2 proteins | Biochemical analysis of BRCA2 function Analysis of BRCA2 function in cells | Often lacks full-length comparison as a control | [77,78,79,80] [76,78] |
| Heterologous expression of cDNA in BRCA2-deficient hamster cells | Variant classification Other functional studies in cells | Inexact conservation between human and hamster BRCA2 | [81,82,83,84] [1,36] |
| BAC expression in mouse embryonic stem cells with floxed Brca2 | Variant classification | Inexact conservation between human and mouse BRCA2 Not very rapid | [67,87,88] |
| piggyBac transposon to transduce human cells with full-length BRCA2 | Variant classification | Low level expression, including for WT BRCA2 | [89] |
| phCMV or BAC for expression of full-length BRCA2 in human cells | In vitro biochemical assays | Levels of expression may not be sufficient for cell-based assays | [1,90] |
| CRISPR-Cas9 mediated gene editing of BRCA2 | Engineering specific BRCA2 mutations in human cells | May not be very rapid Not amenable to generating a protein-wide series of mutations | [91] |
| Specific BRCA2 Function | PALB2-Binding Domain | BRC Repeats | C-Terminal DNA Binding Domain | C-Terminal RAD51-Binding Domain | NLS- 1, 2 and 3 |
|---|---|---|---|---|---|
| BRCA2 Foci | + [8] | ||||
| RAD51 Foci | +/− [78] | + [8] | |||
| HR | +/− [46,76,87] | +/− [78] | + [8,81,82,83,84,87] | − [14,76] | |
| G2 Checkpoint | |||||
| Replication Fork Protection | + [97] | − [14] | + [14] | ||
| Chromosome Stability | +/− [87,97] | +/− [87] | +/− [14] | ||
| Proliferation | + [87] | + [87] | |||
| Resistance to IR | +/− [87] | + [8,81] | |||
| Resistance to Chemotherapeutic Agents and/or MMC | +/− [87] | +/− [78] | + [8,67,81,87] | − [14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreassen, P.R.; Seo, J.; Wiek, C.; Hanenberg, H. Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes 2021, 12, 1034. https://doi.org/10.3390/genes12071034
Andreassen PR, Seo J, Wiek C, Hanenberg H. Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes. 2021; 12(7):1034. https://doi.org/10.3390/genes12071034
Chicago/Turabian StyleAndreassen, Paul R., Joonbae Seo, Constanze Wiek, and Helmut Hanenberg. 2021. "Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses" Genes 12, no. 7: 1034. https://doi.org/10.3390/genes12071034
APA StyleAndreassen, P. R., Seo, J., Wiek, C., & Hanenberg, H. (2021). Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes, 12(7), 1034. https://doi.org/10.3390/genes12071034