
Dissecting the Involvement of Ras GTPases in Kidney Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Kidney Fibrosis
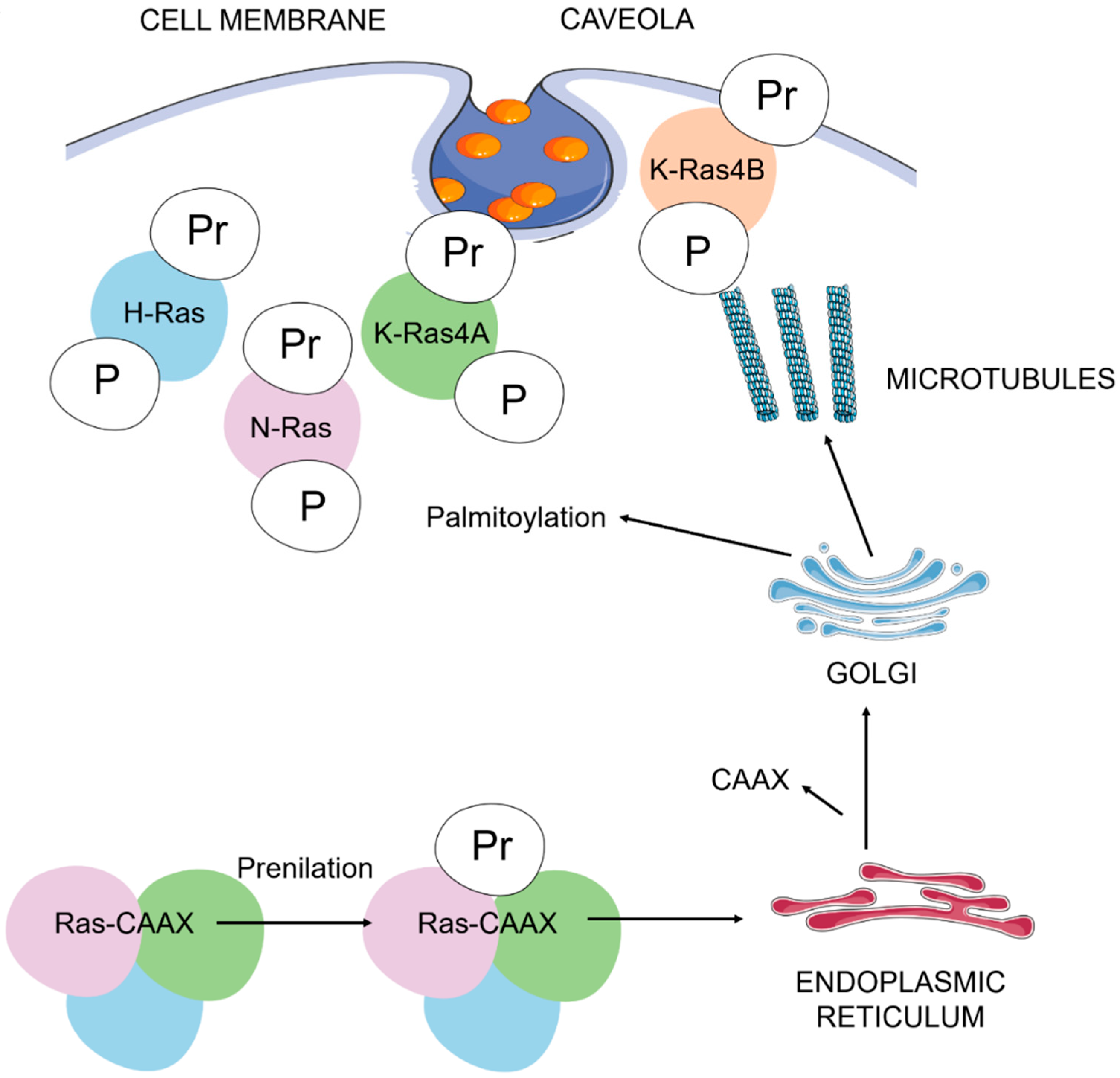
2. Ras Proteins and Their Role in Cell Signalling
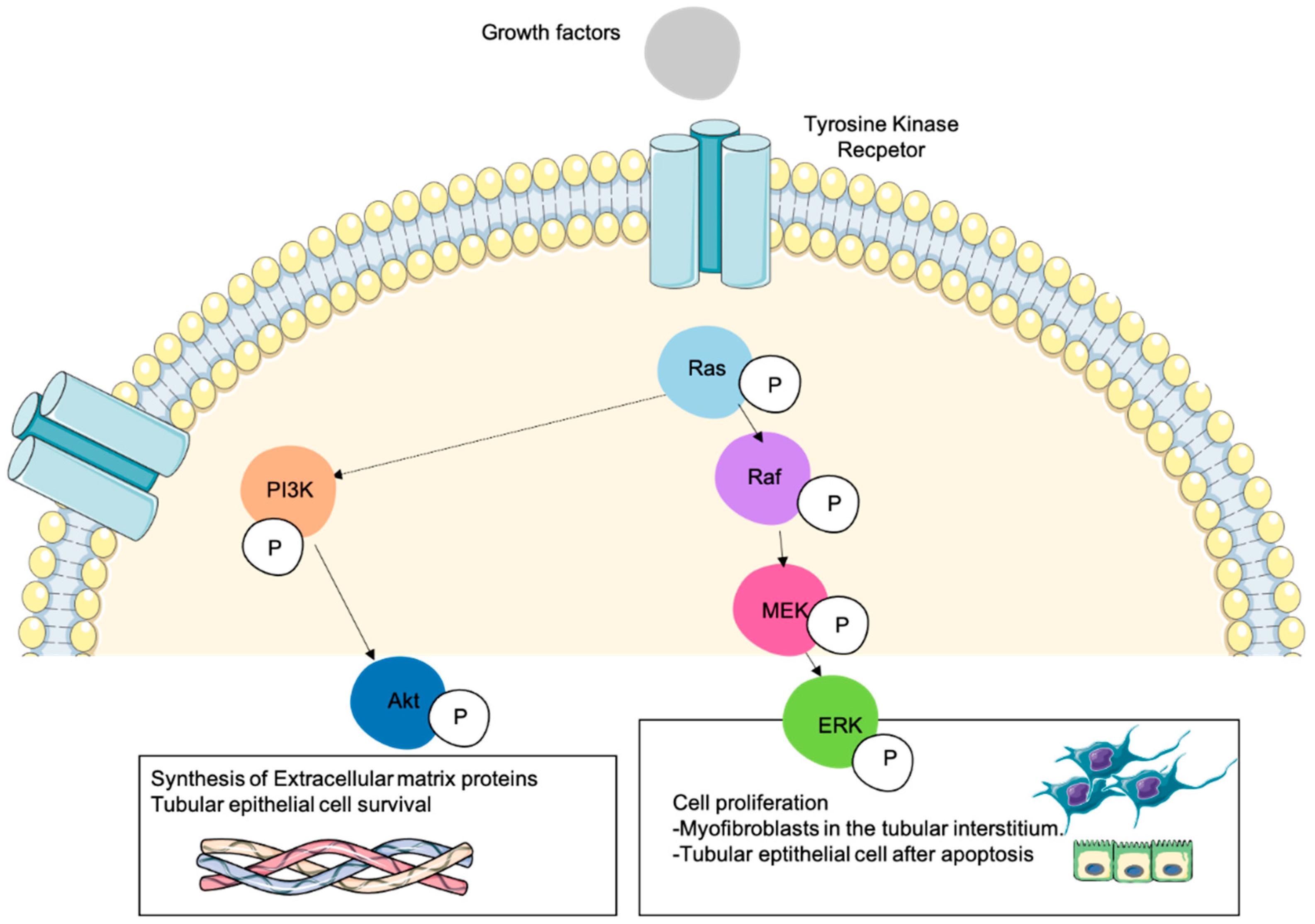
3. Role of Ras Proteins and Ras-Activated Pathways in the Cellular Mechanisms Taking Place in Kidney Fibrosis
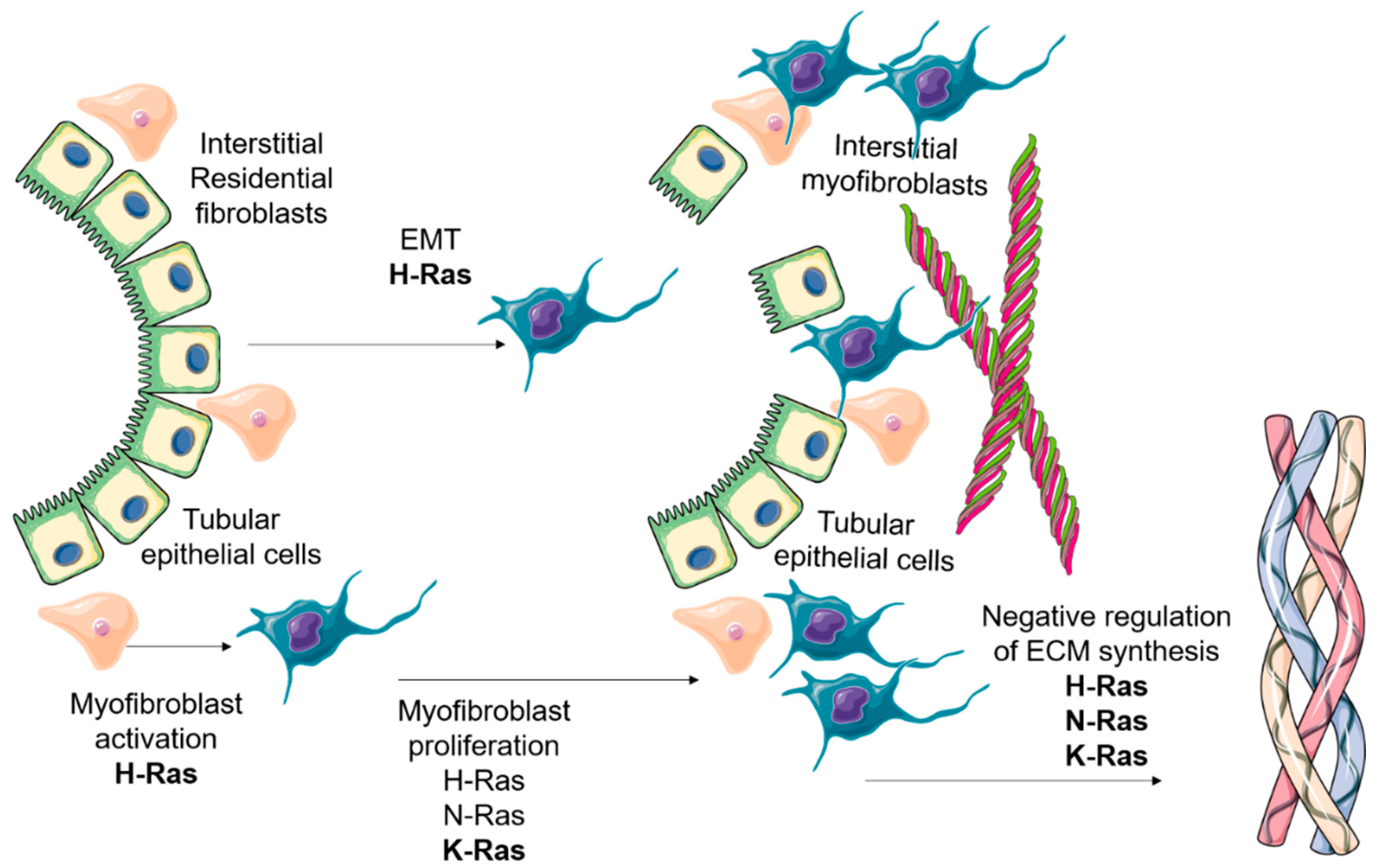
3.1. Effects on Myofibroblast Abundance
3.1.1. Role of Ras Isoforms in the EMT Program in Kidney Fibrosis
3.1.2. Role of Ras Isoforms in the Renal Interstitial Fibroblast Proliferation
3.1.3. Ras Activated Pathways Regulate Cell Proliferation
3.2. Regulation of ECM Protein Synthesis
3.2.1. Ras Proteins as Regulators of ECM Protein Synthesis
3.2.2. Ras Activated Pathways Regulate ECM Deposition
4. Functional Validation of the Role of Ras Isoforms in Kidney Fibrosis Development Using In Vivo Studies
5. Potential Therapeutic Approaches Targeting the Ras Pathway
5.1. Antisense Oligodeoxynucleotides (ASO)
5.2. Inhibition of the Ras Pathway by Inhibiting ‘Upstream’ Activation by Angiotensin II
5.3. Inhibition of the Synthesis of the Farnesyl Group and Farnesylation
5.4. MAP Kinase Inhibitors
5.4.1. Inhibition of the ERK1/2 Pathway
5.4.2. Inhibition of the p38 Pathway
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mutsaers, H.A.M.; Olinga, P. Editorial: Organ Fibrosis: Triggers, Pathways, and Cellular Plasticity. Front. Med. 2016, 3, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. Role of TGF-β in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012, 347, 141–154. [Google Scholar] [CrossRef]
- Grande, M.T.; Perez-Barriocanal, F.; Lopez-Novoa, J.M. Role of inflammation in tubulo-interstitial damage as-sociated to obstructive nephropathy. J. Inflamm. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Kalluri, R. Physiology of the Renal Interstitium. Clin. J. Am. Soc. Nephrol. 2015, 10, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogo, A.B. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Felix, J.M.; Oujo, B.; Lopez-Novoa, J.M. The role of endoglin in kidney fibrosis. Expert Rev. Mol. Med. 2014, 16, e18. [Google Scholar] [CrossRef]
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef]
- Hinz, B. Mechanical aspects of lung fibrosis: A spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 2012, 9, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Picard, N.; Baum, O.; Vogetseder, A.; Kaissling, B.; Le Hir, M. Origin of renal myofibroblasts in the model of uni-lateral ureter obstruction in the rat. Histochem. Cell Biol. 2008, 130, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ Progenitors Are Key Contributors to Injury-Induced Organ Fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Felix, J.M.; Gonzalez-Nunez, M.; Martinez-Salgado, C.; Lopez-Novoa, J.M. TGF-β/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol. Ther. 2015, 156, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sanchez, O.; Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. An integrative view on the role of TGF-β in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010, 77, 950–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Lopez-Novoa, J.M. Fibroblast activation and myofibroblast generation in obstructive nephropa-thy. Nat. Rev. Nephrol. 2009, 5, 319–328. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mecha-nisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Arozarena, I.; Calvo, F.; Crespo, P. Ras, an Actor on Many Stages: Posttranslational Modifications, Localization, and Site-Specified Events. Genes Cancer 2011, 2, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-Binding Proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Fotiadou, P.P.; Takahashi, C.; Rajabi, H.N.; Ewen, M.E. Wild-Type NRas and KRas Perform Distinct Functions during Transformation. Mol. Cell. Biol. 2007, 27, 6742–6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salgado, C.; Rodríguez-Peña, A.B.; Lopez-Novoa, J.M. Involvement of small Ras GTPases and their effectors in chronic renal disease. Cell. Mol. Life Sci. 2008, 65, 477–492. [Google Scholar] [CrossRef]
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004, 14, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. Ras Activation of the Raf Kinase: Tyrosine Kinase Recruitment of the MAP Kinase Cascade. Recent Prog. Horm. Res. 2001, 56, 127–156. [Google Scholar] [CrossRef]
- Hamad, N.M.; Elconin, J.H.; Karnoub, A.E.; Bai, W.; Rich, J.N.; Abraham, R.T.; Der, C.J.; Counter, C.M. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002, 16, 2045–2057. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.F.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and Function of 3-Phosphorylated Inositol Lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Akino, K.; Toyota, M.; Suzuki, H.; Mita, H.; Sasaki, Y.; Ohe-Toyota, M.; Issa, J.-P.J.; Hinoda, Y.; Imai, K.; Tokino, T. The Ras Effector RASSF2 Is a Novel Tumor-Suppressor Gene in Human Colorectal Cancer. Gastroenterology 2005, 129, 156–169. [Google Scholar] [CrossRef]
- Tall, G.G.; Barbieri, M.; Stahl, P.D.; Horazdovsky, B.F. Ras-Activated Endocytosis Is Mediated by the Rab5 Guanine Nucleotide Exchange Activity of RIN1. Dev. Cell 2001, 1, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Marshall, M.S. Ras target proteins in eukaryotic cells. FASEB J. 1995, 9, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Meco, M.T.; Lozano, J.; Municio, M.M.; Berra, E.; Frutos, S.; Sanz, L.; Moscat, J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J. Biol. Chem. 1994, 269, 31706–31710. [Google Scholar] [CrossRef]
- Voice, J.K.; Klemke, R.L.; Le, A.; Jackson, J.H. Four Human Ras Homologs Differ in Their Abilities to Activate Raf-1, Induce Transformation, and Stimulate Cell Motility. J. Biol. Chem. 1999, 274, 17164–17170. [Google Scholar] [CrossRef] [Green Version]
- Vos, M.D.; Ellis, C.A.; Elam, C.; Ülkü, A.S.; Taylor, B.J.; Clark, G.J. RASSF2 Is a Novel K-Ras-specific Effector and Potential Tumor Suppressor. J. Biol. Chem. 2003, 278, 28045–28051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, A.B.; Bar-Sagi, D. Differential Activation of the Rac Pathway by Ha-Ras and K-Ras. J. Biol. Chem. 2001, 276, 15609–15615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, H.M.; Moorhead, J.; Sharpe, C.C.; Dockrell, M.E.C.; Al-Nawab, M.; Hendry, B.M. Expression of Ras GTPases in normal kidney and in glomerulonephritis. Nephrol. Dial. Transplant. 2003, 18, 2284–2292. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Peña, A.B.; Grande, M.T.; Eleno, N.; Arévalo, M.; Guerrero, C.; Santos, E.; López-Novoa, J.M. Activation of Erk1/2 and Akt following unilateral ureteral obstruction. Kidney Int. 2008, 74, 196–209. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.S.; Timms, J.F.; Waterfield, M.D. Cellular Function of Phosphoinositide 3-Kinases: Implications for Development, Immunity, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Hirschel, B.J.; Gabbiani, G.; Ryan, G.B.; Majno, G. Fibroblasts of Granulation Tissue: Immunofluorescent Staining with Antismooth Muscle Serum. Proc. Soc. Exp. Biol. Med. 1971, 138, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Sandbo, N.; Dulin, N. Actin cytoskeleton in myofibroblast differentiation: Ultrastructure defining form and driving function. Transl. Res. 2011, 158, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, N.G.; O’Sullivan, O.E.; Healy, D.A.; Murphy, M.; O’neill, A.J.; Fitzpatrick, J.M.; Watson, R.W.G. TGF-beta1-induced EMT can occur independently of its proapoptotic effects and is aided by EGF receptor activation. Am. J. Physiol. Renal. Physiol. 2006, 290, F1202–F1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Teng, Y.; O’Connell, J.T.; Charytan, D.; Müller, G.A.; Müller, C.A.; Sugimoto, H.; Kalluri, R. Identification of human epididymis protein-4 as a fibroblast-derived mediator of fibrosis. Nat. Med. 2013, 19, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M. Ángela Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.I.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Simon-Tillaux, N.; Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transplant. 2016, 32, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Kaissling, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.-I.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Fuentes-Calvo, I.; Arévalo, M.; Heredia, F.; Santos, E.; Martínez-Salgado, C.; Rodríguez-Puyol, D.; Nieto, M.A.; López-Novoa, J.M. Deletion of H-Ras decreases renal fibrosis and myofibroblast activation following ureteral obstruction in mice. Kidney Int. 2010, 77, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Koesters, R.; Kaissling, B.; Lehir, M.; Picard, N.; Theilig, F.; Gebhardt, R.; Glick, A.B.; Hähnel, B.; Hosser, H.; Gröne, H.-G.; et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am. J. Pathol. 2010, 177, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[β] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.T.; Lee, H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase acti-vation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Schramek, H.; Feifel, E.; Marschitz, I.; Golochtchapova, N.; Gstraunthaler, G.; Montesano, R. Loss of active MEK1-ERK1/2 restores epithelial phenotype and morphogenesis in transdif-ferentiated MDCK cells. Am. J. Physiol. Cell Physiol. 2003, 285, C652–C661. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, C.C.; Dockrell, M.E.C.; Scott, R.; Noor, M.I.; Cowsert, L.M.; Monia, B.P.; Hendry, B.M. Evidence of a Role for Ki-RAS in the Stimulated Proliferation of Renal Fibroblasts. J. Am. Soc. Nephrol. 1999, 10, 1186–1192. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Dockrell, M.E.; Noor, M.I.; Monia, B.P.; Hendry, B.M. Role of Ras isoforms in the stimulated pro-liferation of human renal fibroblasts in primary culture. J. Am. Soc. Nephrol. 2000, 11, 1600–1606. [Google Scholar] [CrossRef]
- Martinez-Salgado, C.; Fuentes-Calvo, I.; Garcia-Cenador, B.; Santos, E.; Lopez-Novoa, J.M. Involvement of H- and N-Ras isoforms in transforming growth factor-beta1-induced proliferation and in collagen and fibronectin synthesis. Exp. Cell Res. 2006, 312, 2093–2106. [Google Scholar] [CrossRef]
- Fuentes-Calvo, I.; Blázquez-Medela, A.M.; Eleno, N.; Santos, E.; Lopez-Novoa, J.M.; Martínez-Salgado, C. H-Ras isoform modulates extracellular matrix synthesis, proliferation, and migration in fibroblasts. Am. J. Physiol. Physiol. 2012, 302, C686–C697. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Calvo, I.; Crespo, P.; Santos, E.; Lopez-Novoa, J.M.; Martinez-Salgado, C. The small GTPase N-Ras regu-lates extracellular matrix synthesis, proliferation and migration in fibroblasts. Biochim. Biophys. Acta 2013, 1833, 2734–2744. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Félix, J.M.; Fuentes-Calvo, I.; Cuesta, C.; Eleno, N.; Crespo, P.; López-Novoa, J.M.; Martínez-Salgado, C. Absence of K-Ras Reduces Proliferation and Migration but Increases Extracellular Ma-trix Synthesis in Fibroblasts. J. Cell Physiol. 2016, 231, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Pat, B.; Yang, T.; Kong, C.; Watters, D.; Johnson, D.W.; Gobe, G. Activation of ERK in renal fibrosis after unilateral ureteral obstruction: Modulation by antioxidants. Kidney Int. 2005, 67, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Foti, R.; Hill, P.A.; Ikezumi, Y.; Atkins, R.C.; Nikolic-Paterson, D.J. Activation of the ERK pathway precedes tubular proliferation in the obstructed rat kidney. Kidney Int. 2003, 63, 1256–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, D.; Guglielmi, K.E.; McGinty, A.; Sorokin, A.; Lianos, E.A.; Dunn, M.J. Activation of extracellular signal-regulated kinase in proliferative glomerulonephritis in rats. J. Clin. Investig. 1997, 100, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, D.; Ostendorf, T.; Kunter UT, A.; Lindemann, M.; Kramer, H.J.; Floege, J. Differential activation of mitogen-activated protein kinases in experimental mesangiopro-liferative glomerulonephritis. J. Am. Soc. Nephrol. 2000, 11, 232–240. [Google Scholar] [CrossRef]
- Bokemeyer, D.; Panek, D.; Kramer, H.J.; Lindemann, M.; Kitahara, M.; Boor, P.; Kerjaschki, D.; Trzaskos, J.M.; Floege, J.; Ostendorf, T. In vivo identification of the mitogen-activated protein kinase cascade as a central patho-genic pathway in experimental mesangioproliferative glomerulonephritis. J. Am. Soc. Nephrol. 2002, 13, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Omori, S.; Hida, M.; Fujita, H.; Takahashi, H.; Tanimura, S.; Kohno, M.; Awazu, M. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycys-tic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.-K.; Cho, W.Y.; Sung, S.A.; Kim, H.K.; Won, N.H. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int. 2005, 67, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-González, P.; Vicente-Sánchez, C.; Pérez-Barriocanal, F.; López-Novoa, J.M.; Morales, A.I. Expres-sion of Ras in cadmium-induced nephrotoxicity. Effects of the natural antioxidant quercetin. Toxicol. Lett. 2006, 164, 112. [Google Scholar] [CrossRef]
- Kahan, C.; Seuwen, K.; Meloche, S.; Pouyssegur, J. Coordinate, biphasic activation of p44 mitogen-activated pro-tein kinase and S6 kinase by growth factors in hamster fibroblasts. Evidence for thrombin-induced signals dif-ferent from phosphoinositide turnover and adenylylcyclase inhibition. J. Biol. Chem. 1992, 267, 13369–13375. [Google Scholar] [CrossRef]
- Qiu, M.S.; Green, S.H. Green, PC12 cell neuronal differentiation is associated with prolonged p21ras activity and con-sequent prolonged ERK activity. Neuron 1992, 9, 705–717. [Google Scholar] [CrossRef]
- Pumiglia, K.M.; Decker, S.J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roovers, K.; Assoian, R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Ishida, T.; Haneda, M.; Maeda, S.; Koya, D.; Kikkawa, R. Stretch-induced overproduction of fibronectin in mesan-gial cells is mediated by the activation of mitogen-activated protein kinase. Diabetes 1999, 48, 595–602. [Google Scholar] [CrossRef]
- Phan, T.-T.; Lim, I.J.; Bay, B.H.; Qi, R.; Longaker, M.T.; Lee, S.-T.; Huynh, H. Role of IGF system of mitogens in the induction of fibroblast proliferation by keloid-derived keratinocytes in vitro. Am. J. Physiol. Cell Physiol. 2003, 284, C860–C869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pat, B.K.; Cuttle, L.; Watters, D.; Yang, T.; Johnson, D.W.; Gobe, G.C. Fibrogenic stresses activate different mitogen-activated protein kinase pathways in renal epi-thelial, endothelial or fibroblast cell populations. Nephrology (Carlton) 2003, 8, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, V.S. Low density lipoproteins and mitogenic signal transduction processes: Role in the pathogenesis of renal disease. Histol. Histopathol. 2002, 17, 497–505. [Google Scholar]
- Kwon, D.S.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, Y.K. Signal transduction of MEK/ERK and PI3K/Akt activation by hypoxia/reoxygenation in renal epithelial cells. Eur. J. Cell Biol. 2006, 85, 1189–1199. [Google Scholar] [CrossRef]
- Arany, I.; Megyesi, J.; Nelkin, B.; Safirstein, R.L. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006, 70, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.D.; Choi, Y.-J.; Tsao, C.C.; Ayala, G.; Sheikh-Hamad, D.; Nassar, G.; Suki, W.N. Renal cell apoptosis in chronic obstructive uropathy: The roles of caspases. Kidney Int. 2001, 60, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, M.; Kase, S.; Adachi, K.; Takeda, A.; Hashimoto, K.; Ito, H. Inhibition of the PI3K-Akt signaling pathway enhances the sensitivity of Fas-mediated apopto-sis in human gastric carcinoma cell line, MKN-45. J. Cancer Res. Clin. Oncol. 2004, 130, 8–14. [Google Scholar] [CrossRef]
- Sinha, D.; Bannergee, S.; Schwartz, J.H.; Lieberthal, W.; Levine, J.S. Inhibition of ligand-independent ERK1/2 activity in kidney proximal tubular cells deprived of soluble survival factors up-regulates Akt and prevents apoptosis. J. Biol. Chem. 2004, 279, 10962–10972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.-K.; Sheu, M.-L.; Hung, K.-Y.; Wu, K.-D.; Liu, S.-H. Honokiol, a small molecular weight natural product, alleviates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2006, 70, 682–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloberas, N.; Cruzado, J.M.; Franquesa, M.; Herrero-Fresneda, I.; Torras, J.; Alperovich, G.; Rama, I.; Vidal, A.; Grinyó, J.M. Mammalian Target of Rapamycin Pathway Blockade Slows Progression of Diabetic Kidney Disease in Rats. J. Am. Soc. Nephrol. 2006, 17, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Nagai, K.; Matsubara, T.; Mima, A.; Sumi, E.; Kanamori, H.; Iehara, N.; Fukatsu, N.; Yanagita, M.; Nakano, T.; Ishimoto, Y.; et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005, 68, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Krepinsky, J.C.; Li, Y.; Chang, Y.; Liu, L.; Peng, F.; Wu, D.; Tang, D.; Scholey, J.; Ingram, A.J. Akt Mediates Mechanical Strain-Induced Collagen Production by Mesangial Cells. J. Am. Soc. Nephrol. 2005, 16, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Zheng, X.; Qin, J.; Chen, Z.; Jin, Y.; Ding, W. Role of PI3-kinase/Akt signalling pathway in renal function and cell proliferation after renal is-chaemia/reperfusion injury in mice. Nephrology (Carlton) 2006, 11, 207–212. [Google Scholar] [CrossRef]
- Sharples, E.J.; Patel, N.; Brown, P.; Stewart, K.; Mota-Philipe, H.; Sheaff, M.; Kieswich, J.; Allen, D.; Harwood, S.; Raftery, M.; et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ische-mia-reperfusion. J. Am. Soc. Nephrol. 2004, 15, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreucci, M.; Michael, A.; Kramers, C.; Park, K.M.; Chen, A.; Matthaeus, T.; Alessandrini, A.; Haq, S.; Force, T.; Bonventre, J.V.; et al. Renal ischemia/reperfusion and ATP depletion/repletion in LLC-PK1 cells result in phosphorylation of FKHR and FKHRL1. Kidney Int. 2003, 64, 1189–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Semenza, G.L. Phosphatidylinositol-3-Kinase Signaling Is Required for Erythropoietin-Mediated Acute Protection against Myocardial Ischemia/Reperfusion Injury. Circulation 2004, 109, 2050–2053. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Dunschede, F.; Koch, E.; Vollmar, A.M.; Kiemer, A.K. α-lipoic acid preconditioning reduces is-chemia-reperfusion injury of the rat liver via the PI3-kinase/Akt pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G769–G778. [Google Scholar] [CrossRef] [Green Version]
- Okumura, H.; Nagaya, N.; Itoh, T.; Okano, I.; Hino, J.; Mori, K.; Tsukamoto, Y.; Ishibashi-Ueda, H.; Miwa, S.; Tambara, K.; et al. Adrenomedullin Infusion Attenuates Myocardial Ischemia/Reperfusion Injury through the Phosphatidylinositol 3-Kinase/Akt-Dependent Pathway. Circulation 2004, 109, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Felix, J.M.; Perretta-Tejedor, N.; Eleno, N.; Lopez-Novoa, J.M.; Martinez-Salgado, C. ALK1 heterozygosi-ty increases extracellular matrix protein expression, proliferation and migration in fibroblasts. Biochim. Biophys. Acta 2014, 1843, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Terada, Y.; Yamada, T.; Takayama, M.; Nonoguchi, H.; Sasaki, S.; Tomita, K.; Marumo, F. Presence and regulation of Raf-1-K (Kinase), MAPK-K, MAP-K, and S6-K in rat nephron segments. J. Am. Soc. Nephrol. 1995, 6, 1565–1577. [Google Scholar] [CrossRef]
- Wisdom, R.; Huynh, L.; Hsia, D.; Kim, S. RAS and TGF-β exert antagonistic effects on extracellular matrix gene expression and fibroblast transformation. Oncogene 2005, 24, 7043–7054. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.H.; Chen, A.; Beno, D.W.A. Raf and Mitogen-activated Protein Kinase Regulate Stellate Cell Collagen Gene Expression. J. Biol. Chem. 1996, 271, 11039–11042. [Google Scholar] [CrossRef] [Green Version]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Casini, A.; Marucci, L.; Gaggiotti, G.; Orlandoni, P.; Macarri, G.; Perego, L.; Benedetti, A.; et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 1999, 29, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Neugarten, J.; Medve, I.; Lei, J.; Silbiger, S.R. Estradiol suppresses mesangial cell type I collagen synthesis via activation of the MAP kinase cascade. Am. J. Physiol. 1999, 277, F875–F881. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Poncelet, A.C.; Hubchak, S.C.; Schnaper, H.W. TGF-beta1 activates MAP kinase in human mesangial cells: A possible role in collagen expression. Kidney Int. 1999, 56, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharaux, P.-L.; Chatziantoniou, C.; Fakhouri, F.; Dussaule, J.-C. Angiotensin II Activates Collagen I Gene through a Mechanism Involving the MAP/ER Kinase Pathway. Hypertension 2000, 36, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reunanen, N.; Foschi, M.; Han, J.; Kahari, V.M. Activation of extracellular signal-regulated kinase 1/2 inhibits type I collagen expression by human skin fibroblasts. J. Biol. Chem. 2000, 275, 34634–34639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Wang, F.S.; Kuo, Y.R.; Huang, Y.T.; Huang, H.C.; Sun, Y.C.; Kuo, Y.H. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabe-tes-induced renal injuries. Kidney Int. 2006, 69, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Stratton, R.; Rajkumar, V.; Ponticos, M.; Nichols, B.; Shiwen, X.; Black, C.M.; Abraham, D.J.; Leask, A. Prostacyclin derivatives prevent the fibrotic response to TGF-β by inhibiting the Ras/MEK/ERK pathway. FASEB J. 2002, 16, 1949–1951. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Blom, I.E.; Sa, S.; Goldschmeding, R.; Abraham, D.J.; Leask, A. CTGF expression in mesangial cells: Involvement of SMADs, MAP kinase, and PKC. Kidney Int. 2002, 62, 1149–1159. [Google Scholar] [CrossRef]
- Phanish, M.K.; Wahab, N.A.; Hendry, B.M.; Dockrell, M.E. TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signalling. Nephron. Exp. Nephrol. 2005, 100, e156. [Google Scholar] [CrossRef]
- Munshi, H.G.; Wu, Y.I.; Mukhopadhyay, S.; Ottaviano, A.J.; Sassano, A.; Koblinski, J.E.; Platanias, L.C.; SharonStack, M. Differential regulation of membrane type 1-matrix metalloproteinase activity by ERK 1/2- and p38 MAPK-modulated tissue inhibitor of metalloproteinases 2 expression controls transforming growth factor-beta1-induced pericellular collagenolysis. J. Biol. Chem. 2004, 279, 39042–39050. [Google Scholar] [CrossRef] [Green Version]
- Coulson, M.T.; Jablonski, P.; Howden, B.O.; Thomson, N.M.; Stein, A.N. Beyond Operational Tolerance: Effect of Ischemic Injury on Development of Chronic Damage in Renal Grafts. Transplantation 2005, 80, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Chen, A.; Bonventre, J.V. Prevention of Kidney Ischemia/Reperfusion-induced Functional Injury and JNK, p38, and MAPK Kinase Activation by Remote Ischemic Pretreatment. J. Biol. Chem. 2001, 276, 11870–11876. [Google Scholar] [CrossRef] [Green Version]
- Park, K.M.; Kramers, C.; Vayssier-Taussat, M.; Chen, A.; Bonventre, J.V. Prevention of kidney ische-mia/reperfusion-induced functional injury, MAPK and MAPK kinase activation, and inflammation by remote transient ureteral obstruction. J. Biol. Chem. 2002, 277, 2040–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Mari, J.F.; Davis, R.; Safirstein, R.L. MAPK activation determines renal epithelial cell survival during oxida-tive injury. Am. J. Physiol. 1999, 277, F195–F203. [Google Scholar] [PubMed]
- Wu, C.-C.; Hung, C.-N.; Shin, Y.-C.; Wang, C.-J.; Huang, H.-P. Myrciaria cauliflora extracts attenuate diabetic nephropathy involving the Ras signaling pathway in streptozotocin/nicotinamide mice on a high fat diet. J. Food Drug Anal. 2016, 24, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Calvo, I.; Martinez-Salgado, C. Sos1 Modulates Extracellular Matrix Synthesis, Proliferation, and Migration in Fibroblasts. Front. Physiol. 2021, 12, 645044. [Google Scholar] [CrossRef]
- Ricupero, D.A.; Poliks, C.F.; Rishikof, D.C.; Cuttle, K.A.; Kuang, P.P.; Goldstein, R.H. Phosphatidylinositol 3-kinase-dependent stabilization of alpha1(I) collagen mRNA in human lung fibroblasts. Am. J. Physiol. Physiol. 2001, 281, C99–C105. [Google Scholar] [CrossRef]
- Ivarsson, M.; McWhirter, A.; Borg, T.K.; Rubin, K. Type I collagen synthesis in cultured human fibroblasts: Regu-lation by cell spreading, platelet-derived growth factor and interactions with collagen fibers. Matrix Biol. 1998, 16, 409–425. [Google Scholar] [CrossRef]
- Grande, M.T.; Arévalo, M.; Nunez, A.; Cannata-Andía, J.B.; Santos, E.; López-Novoa, J.M. Targeted genomic disruption of H-ras and N-ras has no effect on early renal changes after unilateral ureteral ligation. World J. Urol. 2009, 27, 787–797. [Google Scholar] [CrossRef]
- Plowman, S.J.; Williamson, D.J.; O’Sullivan, M.J.; Doig, J.; Ritchie, A.-M.; Harrison, D.J.; Melton, D.W.; Arends, M.J.; Hooper, M.L.; Patek, C.E. While K-ras Is Essential for MouseDevelopment, Expression of the K-ras 4A Splice VariantIsDispensable. Mol. Cell. Biol. 2003, 23, 9245–9250. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-H.; Newbury, L.J.; Knisely, A.; Monia, B.; Hendry, B.M.; Sharpe, C.C. Antisense Knockdown of Kras Inhibits Fibrosis in a Rat Model of Unilateral Ureteric Obstruction. Am. J. Pathol. 2012, 180, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Newbury, L.J.; Wang, J.-H.; Hung, G.; Hendry, B.M.; Sharpe, C.C. Inhibition of Kirsten-Ras reduces fibrosis and protects against renal dysfunction in a mouse model of chronic folic acid nephropathy. Sci. Rep. 2019, 9, 14010. [Google Scholar] [CrossRef]
- Wang, J.-H.; Hendry, B.M.; Sharpe, C.C. Silencing genes in the kidney: Antisense or RNA interference? Nephrol. Dial. Transplant. 2008, 23, 2115–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Peña, A.B.; Fuentes-Calvo, I.; Docherty, N.G.; Arévalo, M.; Grande, M.T.; Eleno, N.; Pérez-Barriocanal, F.; Lopez-Novoa, J.M. Effect of Angiotensin II and Small GTPase Ras Signaling Pathway Inhibition on Early Renal Changes in a Murine Model of Obstructive Nephropathy. BioMed Res. Int. 2014, 2014, 124902. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, A.; Santillo, M.; Cicale, M.; Seru, R.; Cuda, G.; Anrather, J.; Iadecola, C.; Postiglione, A.; Avvedimento, E.V.; Paternò, R. Farnesyl transferase inhibitors induce neuroprotection by inhibiting Ha-Ras signalling pathway. Eur. J. Neurosci. 2007, 26, 3261–3266. [Google Scholar] [CrossRef]
- Nogueira, A.; Peixoto, F.; Oliveira, M.M.; Pires, C.A.; Colaço, B.; Oliveira, P.A.; Pires, M.J. The Effects of Long-Term Chaetomellic Acid A Administration on Renal Function and Oxidative Stress in a Rat Model of Renal Mass Reduction. Biomed. Res. Int. 2017, 2017, 5125980. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Vala, H.; Vasconcelos-Nóbrega, C.; Faustino-Rocha, A.I.; Pires, C.A.; Colaço, A.; Oliveira, P.A.; Pires, M.J. Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolo-sclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 2017, 96, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haneda, M.; Araki, S.I.; Togawa, M.; Sugimoto, T.; Motohide, I.; Kikkawa, R. Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glo-merular mesangial cells cultured under high glucose conditions. Diabetes 1997, 46, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Kieswich, J.; Pacheco, S.; Nadarajah, L.; Harwood, S.M.; O’Riordan, C.E.; Thiemermann, C.; Yaqoob, M.M. The MEK Inhibitor Trametinib Ameliorates Kidney Fibrosis by Suppressing ERK1/2 and mTORC1 Signaling. J. Am. Soc. Nephrol. 2019, 30, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, N.; Kohno, M.; Yokoyama, T. Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model. Nephrol. Dial. Transplant. 2012, 27, 1351–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Félix, J.M.; Martínez-Salgado, C. Dissecting the Involvement of Ras GTPases in Kidney Fibrosis. Genes 2021, 12, 800. https://doi.org/10.3390/genes12060800
Muñoz-Félix JM, Martínez-Salgado C. Dissecting the Involvement of Ras GTPases in Kidney Fibrosis. Genes. 2021; 12(6):800. https://doi.org/10.3390/genes12060800
Chicago/Turabian StyleMuñoz-Félix, José M., and Carlos Martínez-Salgado. 2021. "Dissecting the Involvement of Ras GTPases in Kidney Fibrosis" Genes 12, no. 6: 800. https://doi.org/10.3390/genes12060800
APA StyleMuñoz-Félix, J. M., & Martínez-Salgado, C. (2021). Dissecting the Involvement of Ras GTPases in Kidney Fibrosis. Genes, 12(6), 800. https://doi.org/10.3390/genes12060800