
DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML)

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Samples
2.2. DNA Methylation Assay
2.3. Data Preprocessing
2.4. Subtype Prediction with DNA Methylation
2.5. Survival Analysis
2.6. Data Visualization
3. Results
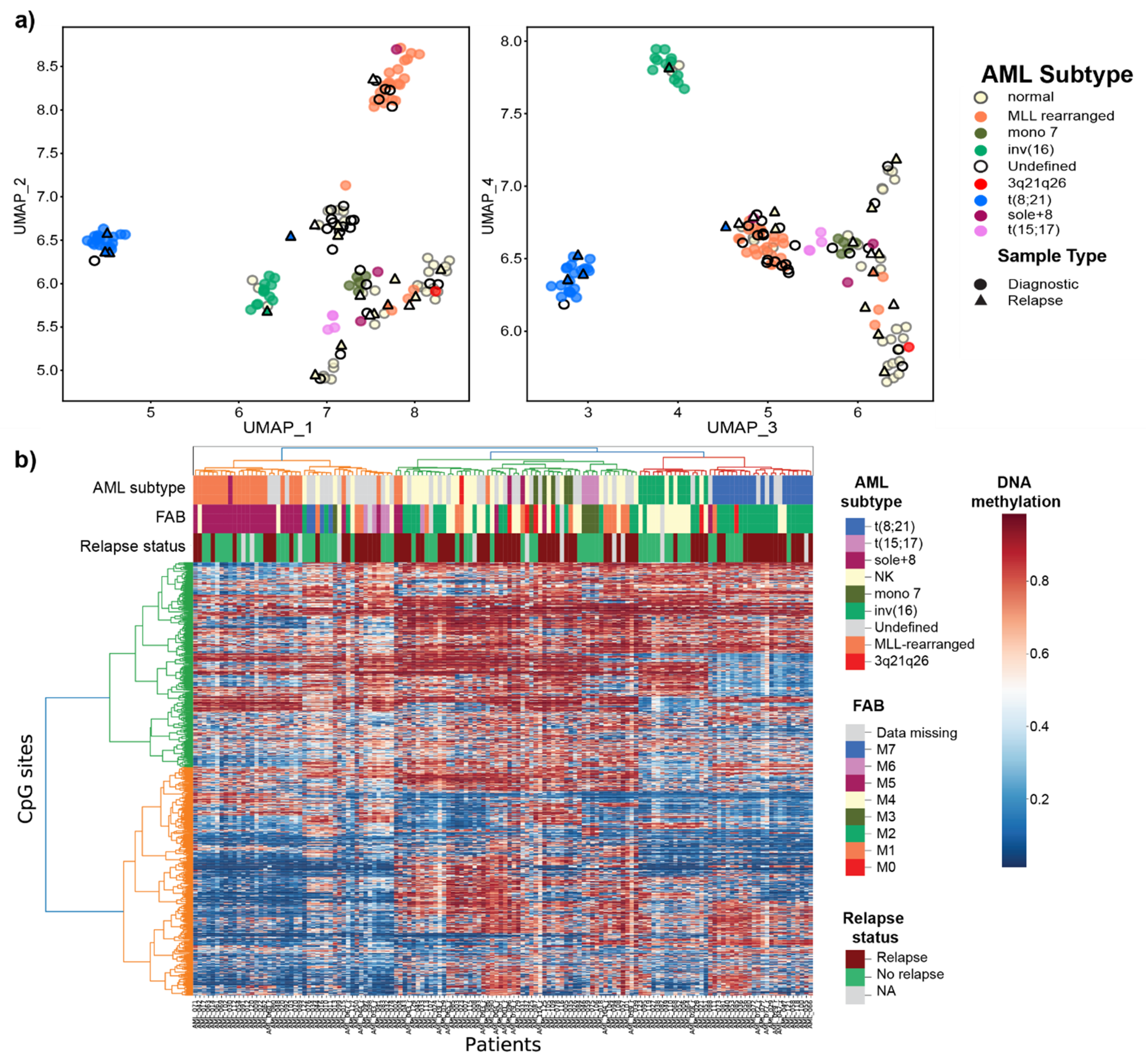
3.1. Overview of the DNA Methylome in Pediatric AML
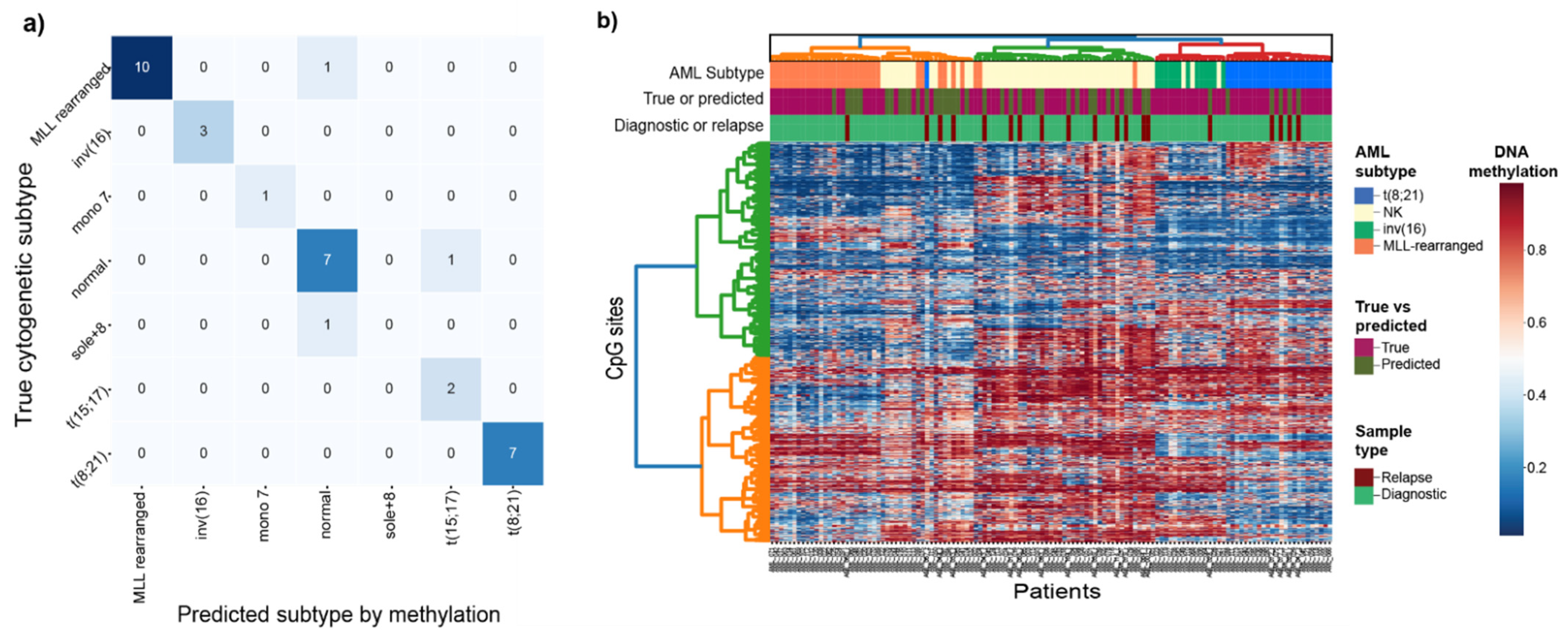
3.2. Prediction of Cytogenetic Subtype with DNA Methylation
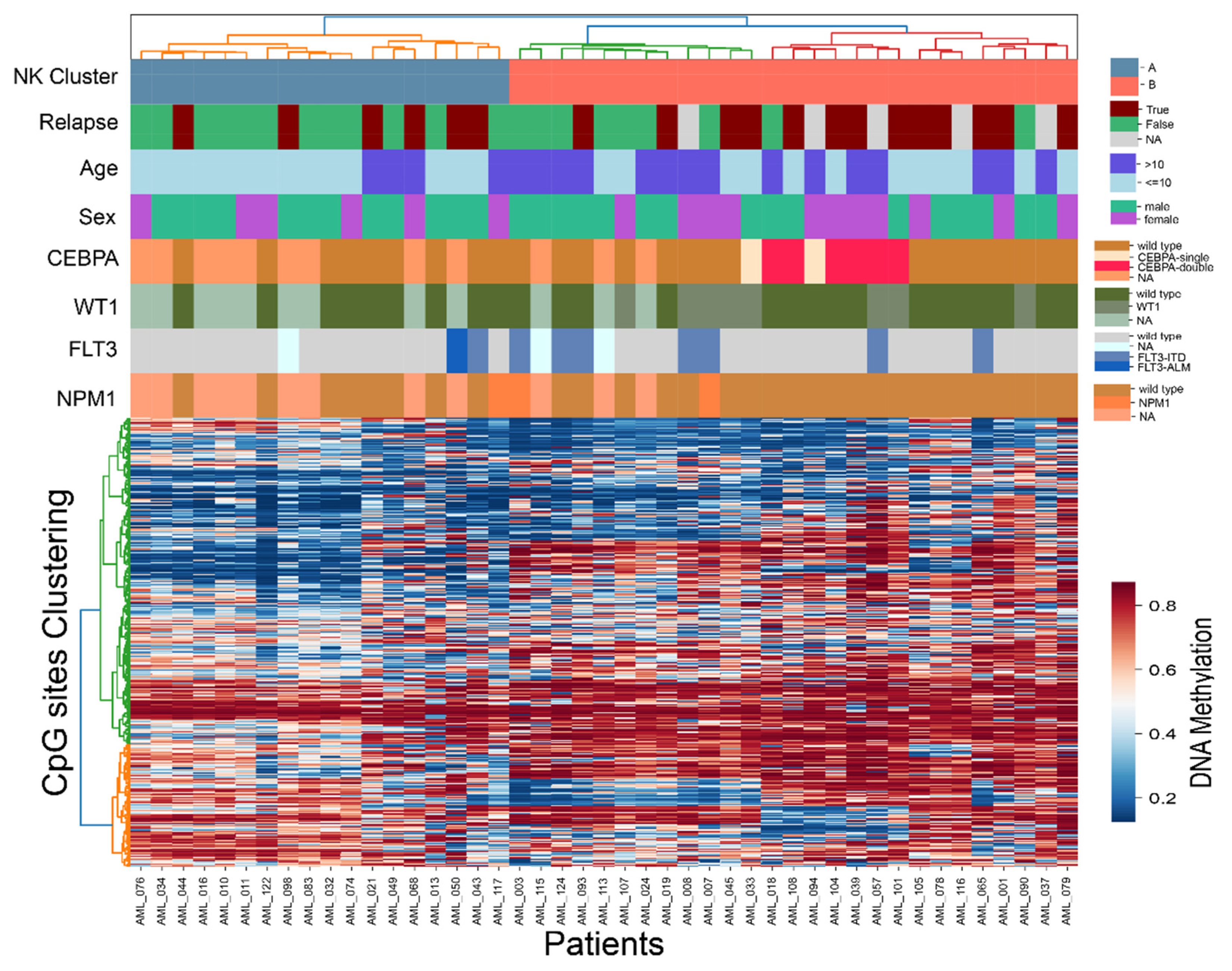
3.3. Intra-Subtype Heterogenetity in NK-AML
3.4. Cytogenetic-Specific DNA Methylation Signatures
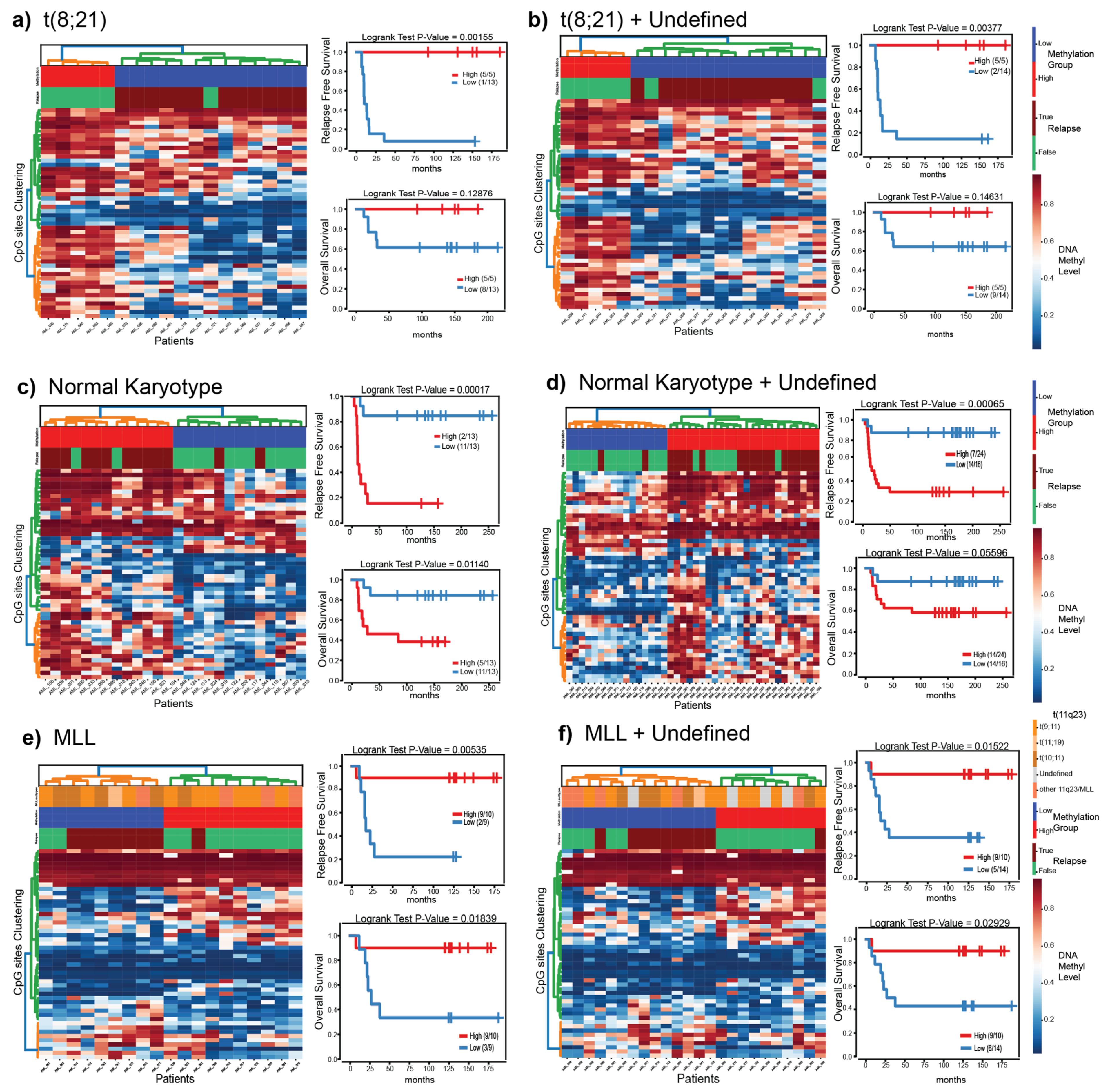
3.5. Putatively Prognostic DNA Methylation Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Ha, S.-Y.; Heldrup, J.; Jahnukainen, K.; Jónsson, Ó.G.; Lausen, B.; Palle, J.; Zeller, B.; et al. The applicability of the WHO classification in paediatric AML. A NOPHO-AML study. Br. J. Haematol. 2015, 169, 859–867. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Dahl, G.; Ribeiro, R.C.; Bowman, W.P.; Taub, J.; Pounds, S.; Razzouk, B.I.; Lacayo, N.J.; Cao, X.; et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: Results of the AML02 multicentre trial. Lancet Oncol. 2010, 11, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Coustan-Smith, E.; Campana, D. Should evaluation for minimal residual disease be routine in acute myeloid leukemia? Curr. Opin. Hematol. 2013, 20, 86–92. [Google Scholar] [CrossRef]
- Manola, K.N. Cytogenetics of pediatric acute myeloid leukemia. Eur. J. Haematol. 2009, 83, 391–405. [Google Scholar] [CrossRef]
- Hollink, I.H.I.M.; Zwaan, C.M.; Zimmermann, M.; Arentsen-Peters, T.C.J.M.; Pieters, R.; Cloos, J.; Kaspers, G.J.L.; de Graaf, S.S.N.; Harbott, J.; Creutzig, U.; et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 2009, 23, 262–270. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Paschka, P.; Whitman, S.P.; Bloomfield, C.D. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification? Blood 2007, 109, 431–448. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Garrett-Bakelman, F.E.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.A.; Huang, Y.; Biswas, D.; Hess, J.L.; et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Gebhard, C.; Glatz, D.; Schwarzfischer, L.; Wimmer, J.; Stasik, S.; Nuetzel, M.; Heudobler, D.; Andreesen, R.; Ehninger, G.; Thiede, C.; et al. Profiling of aberrant DNA methylation in acute myeloid leukemia reveals subclasses of CG-rich regions with epigenetic or genetic association. Leukemia 2019, 33, 26–36. [Google Scholar] [CrossRef]
- Saied, M.H.; Marzec, J.; Khalid, S.; Smith, P.; Down, T.A.; Rakyan, V.K.; Molloy, G.; Raghavan, M.; Debernardi, S.; Young, B.D. Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS ONE 2012, 7, e33213. [Google Scholar]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratmann, S.; Yones, S.A.; Mayrhofer, M.; Norgren, N.; Skaftason, A.; Sun, J.; Smolinska, K.; Komorowski, J.; Herlin, M.K.; Sundstrom, C.; et al. Genomic characterization of relapsed acute myeloid leukemia reveals novel putative therapeutic targets. Blood Adv. 2021, 5, 900–912. [Google Scholar] [CrossRef]
- Juhl-Christensen, C.; Ommen, H.B.; Aggerholm, A.; Lausen, B.; Kjeldsen, E.; Hasle, H.; Hokland, P. Genetic and epigenetic similarities and differences between childhood and adult AML. Pediatric Blood Cancer 2012, 58, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Cao, X.; Raimondi, S.; Downing, J.; Ribeiro, R.; Gruber, T.A.; Rubnitz, J.; Pounds, S. DNA Methylation Clusters and Their Relation to Cytogenetic Features in Pediatric AML. Cancers 2020, 12, 3024. [Google Scholar] [CrossRef] [PubMed]
- Koldobskiy, M.A.; Abante, J.; Jenkinson, G.; Pujadas, E.; Tetens, A.; Zhao, F.; Tryggvadottir, R.; Idrizi, A.; Reinisch, A.; Majeti, R.; et al. A Dysregulated DNA Methylation Landscape Linked to Gene Expression in MLL-Rearranged AML. Epigenetics 2020, 15, 841–858. [Google Scholar] [CrossRef]
- Larmonie, N.S.D.; Arentsen-Peters, T.C.J.M.; Obulkasim, A.; Valerio, D.; Sonneveld, E.; Danen-van Oorschot, A.A.; de Haas, V.; Reinhardt, D.; Zimmermann, M.; Trka, J.; et al. MN1 overexpression is driven by loss of DNMT3B methylation activity in inv(16) pediatric AML. Oncogene 2018, 37, 107–115. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, L.; Lv, N.; Liu, J.; Li, Y.; Chen, X.; Xu, Q.; Chen, G.; Pang, B.; Wang, L.; et al. Methylation-associated silencing of BASP1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zampini, M.; Tregnago, C.; Bisio, V.; Simula, L.; Borella, G.; Manara, E.; Zanon, C.; Zonta, F.; Serafin, V.; Accordi, B.; et al. Epigenetic heterogeneity affects the risk of relapse in children with t(8;21)RUNX1-RUNX1T1-rearranged AML. Leukemia 2018, 32, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Cao, X.; Raimondi, S.C.; Rafiee, R.; Downing, J.R.; Lei, S.; Gruber, T.; Ribeiro, R.C.; Rubnitz, J.E.; Pounds, S.B. Integrated epigenetic and genetic analysis identifies markers of prognostic significance in pediatric acute myeloid leukemia. Oncotarget 2018, 9, 26711–26723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lie, S.O.; Abrahamsson, J.; Clausen, N.; Forestier, E.; Hasle, H.; Hovi, L.; Jonmundsson, G.; Mellander, L.; Gustafsson, G. Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down’s syndrome: Results of NOPHO-AML trials. Br. J. Haematol. 2003, 122, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, J.; Forestier, E.; Heldrup, J.; Jahnukainen, K.; Jonsson, O.G.; Lausen, B.; Palle, J.; Zeller, B.; Hasle, H. Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, G.; Drake, F.L. The Python Language Reference Manual; Network Theory Ltd.: Bristol, UK, 2011. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S. Jupyter Notebooks–A Publishing Format For Reproducible Computational Workflows; IOS Press: Amsterdam The Netherlands, 2016. [Google Scholar]
- Nordlund, J.; Backlin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [Green Version]
- Behdenna, A.; Haziza, J.; Azencott, C.-A.; Nordor, A. pyComBat, a Python tool for batch effects correction in high-throughput molecular data using empirical Bayes methods. BioRxiv 2020. [Google Scholar] [CrossRef]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 2014, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D graphics environment. IEEE Ann. Hist. Comput. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:preprint/1802.03426. Available online: https://umap-learn.readthedocs.io/en/latest/ (accessed on 19 February 2021).
- Staffas, A.; Kanduri, M.; Hovland, R.; Rosenquist, R.; Ommen, H.B.; Abrahamsson, J.; Forestier, E.; Jahnukainen, K.; Jónsson, Ó.G.; Zeller, B. Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2011, 118, 5905–5913. [Google Scholar] [CrossRef]
- Wang, J.; Muntean, A.G.; Hess, J.L. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood J. Am. Soc. Hematol. 2012, 119, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.-J.; Milne, T.A. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Nordlund, J.; Bäcklin, C.L.; Zachariadis, V.; Cavelier, L.; Dahlberg, J.; Öfverholm, I.; Barbany, G.; Nordgren, A.; Övernäs, E.; Abrahamsson, J. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenet. 2015, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; Von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Duran-Ferrer, M.; Clot, G.; Nadeu, F.; Beekman, R.; Baumann, T.; Nordlund, J.; Marincevic-Zuniga, Y.; Lönnerholm, G.; Rivas-Delgado, A.; Martin, S. The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome. Nat. Cancer 2020, 1, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Ehrich, M.; Dohner, K.; Schlenk, R.F.; Dohner, H.; Nelson, M.R.; van den Boom, D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood 2010, 115, 636–642. [Google Scholar] [CrossRef]
- Kelly, A.D.; Kroeger, H.; Yamazaki, J.; Taby, R.; Neumann, F.; Yu, S.; Lee, J.T.; Patel, B.; Li, Y.; He, R.; et al. A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia 2017, 31, 2011–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamato, G.; Yamaguchi, H.; Handa, H.; Shiba, N.; Kawamura, M.; Wakita, S.; Inokuchi, K.; Hara, Y.; Ohki, K.; Okubo, J. Clinical features and prognostic impact of PRDM16 expression in adult acute myeloid leukemia. Genes Chromosomes Cancer 2017, 56, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, J.; Lee, S.Y.; Xiong, J.; Bhanu, N.; Guo, Q.; Ma, P.; Sun, Y.; Rao, R.C.; Garcia, B.A. PRDM16 suppresses MLL1r leukemia via intrinsic histone methyltransferase activity. Mol. Cell 2016, 62, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-Y.; Lin, P.-M.; Hsiao, H.-H.; Hsu, J.-F.; Lin, H.Y.-H.; Hsu, C.-M.; Chen, I.-Y.; Su, S.-W.; Liu, Y.-C.; Lin, S.-F. Up-regulation of PER3 expression is correlated with better clinical outcome in acute leukemia. Anticancer. Res. 2015, 35, 6615–6622. [Google Scholar]
- Simmons, H.M.; Ruis, B.L.; Kapoor, M.; Hudacek, A.W.; Conklin, K.F. Identification of NOM1, a nucleolar, eIF4A binding protein encoded within the chromosome 7q36 breakpoint region targeted in cases of pediatric acute myeloid leukemia. Gene 2005, 347, 137–145. [Google Scholar] [CrossRef]
- Fan, H.; Lu, J.; Guo, Y.; Li, D.; Zhang, Z.-M.; Tsai, Y.-H.; Pi, W.-C.; Ahn, J.H.; Gong, W.; Xiang, Y. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet. 2020, 52, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Harder, L.; Eschenburg, G.; Zech, A.; Kriebitzsch, N.; Otto, B.; Streichert, T.; Behlich, A.-S.; Dierck, K.; Klingler, B.; Hansen, A. Aberrant ZNF423 impedes B cell differentiation and is linked to adverse outcome of ETV6-RUNX1 negative B precursor acute lymphoblastic leukemia. J. Exp. Med. 2013, 210, 2289–2304. [Google Scholar] [CrossRef] [Green Version]
- Batista, I.d.A.A.; Helguero, L.A. Biological processes and signal transduction pathways regulated by the protein methyltransferase SETD7 and their significance in cancer. Signal Transduct. Target. Ther. 2018, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Song, G.; Song, G.; Li, R.; Gao, M.; Ye, L.; Zhang, C. Identification of DNA methylation prognostic signature of acute myelocytic leukemia. PLoS ONE 2018, 13, e0199689. [Google Scholar] [CrossRef] [PubMed]
- Radtke, I.; Mullighan, C.G.; Ishii, M.; Su, X.; Cheng, J.; Ma, J.; Ganti, R.; Cai, Z.; Goorha, S.; Pounds, S.B. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 12944–12949. [Google Scholar] [CrossRef] [Green Version]
- Touzart, A.; Mayakonda, A.; Smith, C.; Hey, J.; Toth, R.; Cieslak, A.; Andrieu, G.P.; Tran Quang, C.; Latiri, M.; Ghysdael, J.; et al. Epigenetic analysis of patients with T-ALL identifies poor outcomes and a hypomethylating agent-responsive subgroup. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef]
- Newcombe, A.A.; Gibson, B.E.; Keeshan, K. Harnessing the potential of epigenetic therapies for childhood acute myeloid leukemia. Exp. Hematol. 2018, 63, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytogenetic Subtype | Normal Karyotype (NK) | MLL/KMT2A Rearranged | Undefined | t(8;21) RUNX1/RUNX1T1 | inv(16) CBFB/MYH11 | mono 7 | t(15;17) PML-RARA | sole +8 | 3q21q26 |
|---|---|---|---|---|---|---|---|---|---|
| Number of diagnostic Samples | 30 | 25 | 24 | 19 | 12 | 5 | 4 | 3 | 1 |
| Number of relapse samples | 8 | 1 | 5 | 4 | 1 | - | - | - | - |
| Age range | 1–18 | 0–16 | 0–17 | 2–16 | 1–17 | 1–5 | 3–16 | 3–14 | 14 |
| CNS involvement | - | - | - | - | - | - | - | - | - |
| Yes | 4 | 1 | 2 | 6 | - | - | - | - | - |
| No | 34 | 25 | 27 | 17 | 12 | 4 | 3 | 3 | 1 |
| Missing | - | - | - | - | 1 | 1 | 1 | - | - |
| SCT | - | - | - | - | - | - | - | - | - |
| Yes | 8 | 3 | 1 | - | - | 5 | - | 2 | - |
| No | 30 | 23 | 27 | 23 | 13 | - | 3 | 1 | 1 |
| Missing | - | - | 1 | - | - | - | 1 | - | - |
| FAB | - | - | - | - | - | - | - | - | - |
| M0 | 2 | - | - | 1 | - | 2 | - | 1 | - |
| M1 | 7 | - | 3 | 1 | - | 1 | - | 1 | 1 |
| M2 | 14 | 1 | 3 | 19 | 3 | 1 | - | - | - |
| M3 | 1 | - | 1 | - | - | - | 4 | - | - |
| M4 | 10 | 1 | 10 | 2 | 9 | - | - | - | - |
| M5 | - | 24 | 4 | - | - | 1 | - | 1 | - |
| M6 | 3 | - | 2 | - | - | - | - | - | - |
| M7 | - | - | 4 | - | - | - | - | - | - |
| Missing | 1 | - | 2 | - | 1 | - | - | - | - |
| Precision | Recall | F1 Score | Total samples Test Set | Total Samples Train Set | |
|---|---|---|---|---|---|
| MLL/KMT2A-rearranged | 1 | 0.91 | 0.95 | 11 | 14 |
| inv(16)/CBFB-MYH11 | 1 | 1 | 1 | 3 | 9 |
| Normal Karyotype (NK) | 0.78 | 0.88 | 0.82 | 8 | 22 |
| t(8;21)/RUNX1-RUNX1T1 | 1 | 1 | 1 | 7 | 12 |
| Subtype | N CpG Sites (Adjusted p Value < 0.05) | N CpG Sites Unique to Subtype (N Genes) | Gene Names (CpG IDs) Unique to Subtype |
|---|---|---|---|
| Normal Karyotype | 569 | 6 (5) | ZNF793 (cg15139588), APBA2 (cg15605858), PRDM16 (cg02390319), PPP1R14A (cg02571816), ACCN1 (cg03745383) |
| MLL/KMT2A-rearranged | 873 | 59 (33) | KIAA1755 (cg14003035), PLAUR (cg27340480), PER3 (cg05803631), ASB2 (cg09341793), KLK4 (cg26827876), BARHL2 (cg18322569), L1TD1 (cg23049458), ARPC1B (cg10428938), ST8SIA6 (cg17256364), NKX6-2 (cg11174855), WNT5A (cg19554389), HOXA5 (cg12128839, cg25307665), NFIX (cg06744585), SNED1 (cg25241559, cg09991306), TNXB (cg12694372, cg10923662, cg16834823, cg01992382), MSX2 (cg06013117), MAPK8IP1 (cg08214808), BNIP3 (cg18477674), CASR (cg19108881), HECW1 (cg24384918), PCDHA1; PCDHA2; PCDHA3; PCDHA4; PCDHA5; PCDHA6; PCDHA7; PCDHA8 (cg19596110), PITX1 (cg00396667), KCNN1 (cg07857792), TMEM132D (cg20168964), NPSR1 (cg20276677), LOC732275 (cg16709904), NOM1 (cg02413092), SPEG (cg16440561), EDARADD (cg09164898), THBS4 (cg26286839), HOOK2 (cg06417478), LOC254559 (cg09969277), DCC (cg25204852) |
| mono7 | 330 | 24 (12) | BCL2 (cg25059899), ZNF577 (cg03562414, cg24794228, cg11269599, cg10635122), ZNF154 (cg21790626, cg27049766, cg26465391), ARRB2 (cg07971820, cg02286380), SKI (cg25139649), ERCC3 (cg06373940), RPTOR (cg09929238), FBXO47 (cg04120272), DLL1 (cg00084338), C1orf86; LOC100128003 (cg26227225), CYP1A1 (cg22549041), PLD6 (cg24578857, cg19093370) |
| inv(16)/CBFB-MYH11 | 571 | 22 (15) | DPF3 (cg13588403), IFLTD1 (cg13134916), BAHCC1 (cg06636541), LEPR (cg16987305), AFAP1 (cg22079161), PRHOXNB (cg20101529), ANK1 (cg19537719), SHISA6 (cg13330559), PRDM16 (cg03337482), MUC4 (cg05834845), C22orf34 (cg20744362), LY96 (cg23732024), ZNF423 (cg26929700, cg04086531), GNG7 (cg26988138), CNTD2 (cg08871608) |
| t(8;21)/RUNX1-RUNX1T1 | 723 | 27 (15) | SMTNL2 (cg13375589), TUSC1 (cg13811417), PDLIM3 (cg14632696), CYP27C1 (cg08022717), PCDHA1; PCDHA10; PCDHA11; PCDHA2; PCDHA3; PCDHA4; PCDHA5; PCDHA6; PCDHA7; PCDHA8; PCDHA9 (cg26514430), RYR2 (cg07790615), TACSTD2 (cg13443627), FBXL7 (cg26134895), VSTM2A (cg19868631), MARCH11 (cg25092681, cg01791874,cg00339556, cg16150752, cg17712694), IGSF21 (cg15564444), TTBK1 (cg16620382), SHROOM1 (cg21811204), ELOVL4 (cg04107099), SETDB1 (cg15448220) |
| t(15;17)/PML-RARA | 328 | 8 (8) | NFYC (cg16167741), C7orf50 (cg23657099), IGDCC4 (cg00776960), SETD7 (cg02409722), SCHIP1 (cg23553912), SYNE1 (cg02796568), WBSCR17 (cg02300154), C21orf7 (cg08854834) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krali, O.; Palle, J.; Bäcklin, C.L.; Abrahamsson, J.; Norén-Nyström, U.; Hasle, H.; Jahnukainen, K.; Jónsson, Ó.G.; Hovland, R.; Lausen, B.; et al. DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML). Genes 2021, 12, 895. https://doi.org/10.3390/genes12060895
Krali O, Palle J, Bäcklin CL, Abrahamsson J, Norén-Nyström U, Hasle H, Jahnukainen K, Jónsson ÓG, Hovland R, Lausen B, et al. DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML). Genes. 2021; 12(6):895. https://doi.org/10.3390/genes12060895
Chicago/Turabian StyleKrali, Olga, Josefine Palle, Christofer L. Bäcklin, Jonas Abrahamsson, Ulrika Norén-Nyström, Henrik Hasle, Kirsi Jahnukainen, Ólafur Gísli Jónsson, Randi Hovland, Birgitte Lausen, and et al. 2021. "DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML)" Genes 12, no. 6: 895. https://doi.org/10.3390/genes12060895
APA StyleKrali, O., Palle, J., Bäcklin, C. L., Abrahamsson, J., Norén-Nyström, U., Hasle, H., Jahnukainen, K., Jónsson, Ó. G., Hovland, R., Lausen, B., Larsson, R., Palmqvist, L., Staffas, A., Zeller, B., & Nordlund, J. (2021). DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML). Genes, 12(6), 895. https://doi.org/10.3390/genes12060895