
Comprehensive Analysis of RNA Expression Correlations between Biofluids and Human Tissues




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
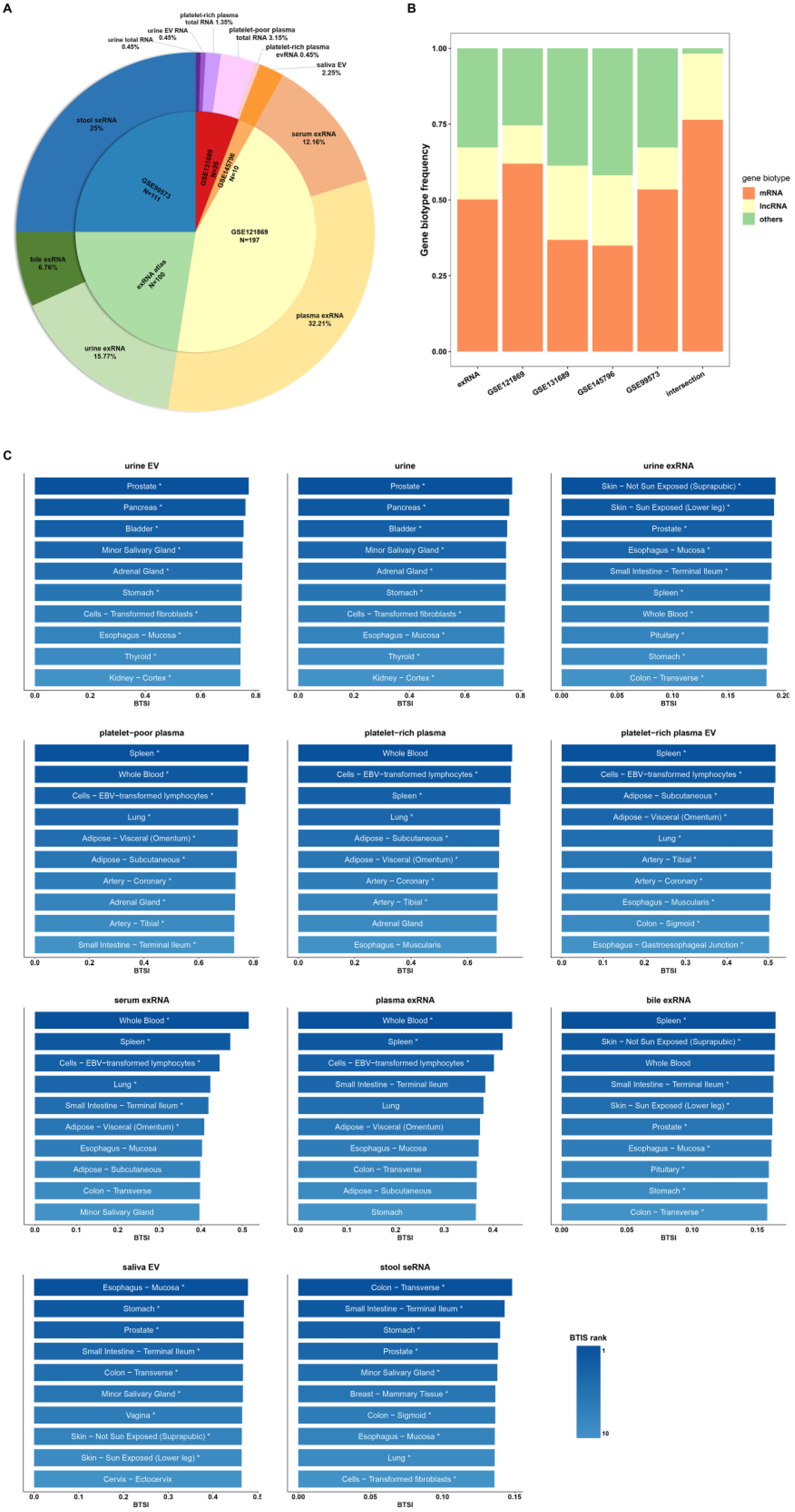
2.1. Data Collection
2.2. Data Preprocess
2.3. Statistical Analysis
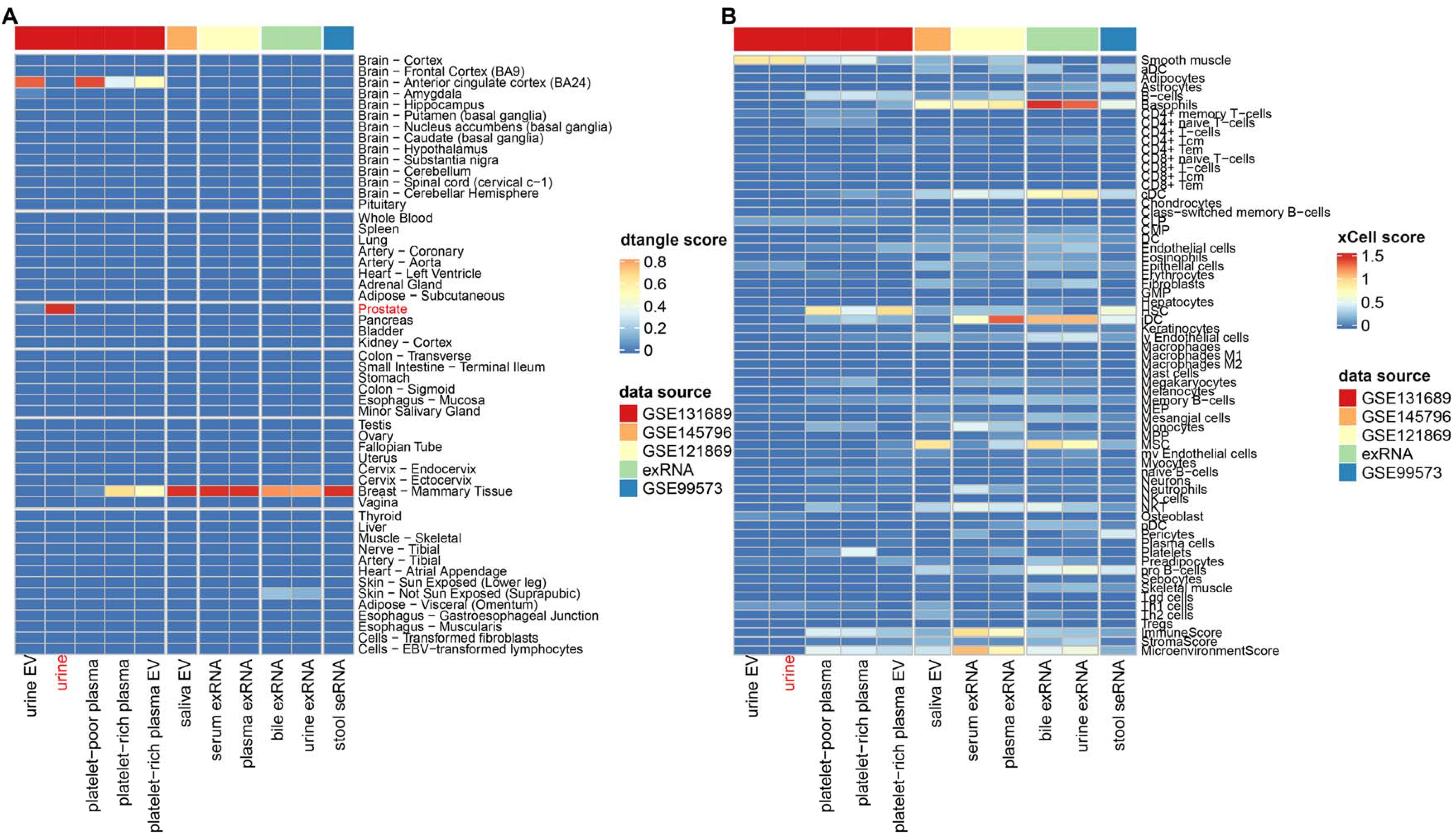
2.4. Decomposition of Tissue Component in Biofluid
2.5. Functional Enrichment Analysis of Stable and Variable Components of Biofluid Genome
3. Results
3.1. Biofluids and Tissues Exhibit Widely Positive Correlations
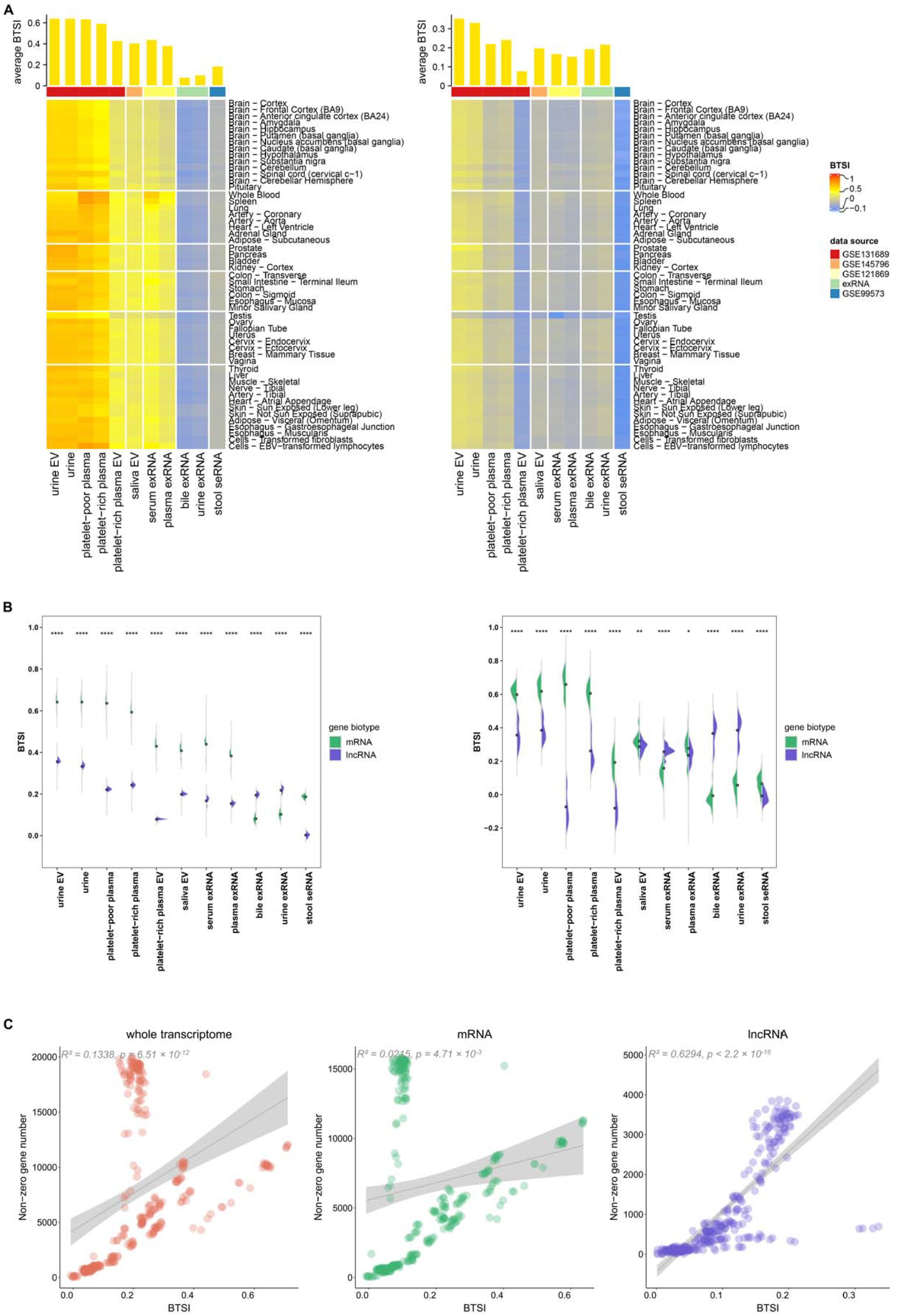
3.2. Tendency and Specificity of Biofluid–Tissue Correlation
3.3. Biofluid-Derived mRNAs Show Stronger Correlation with Healthy Tissues Than Biofluid-Derived lncRNAs
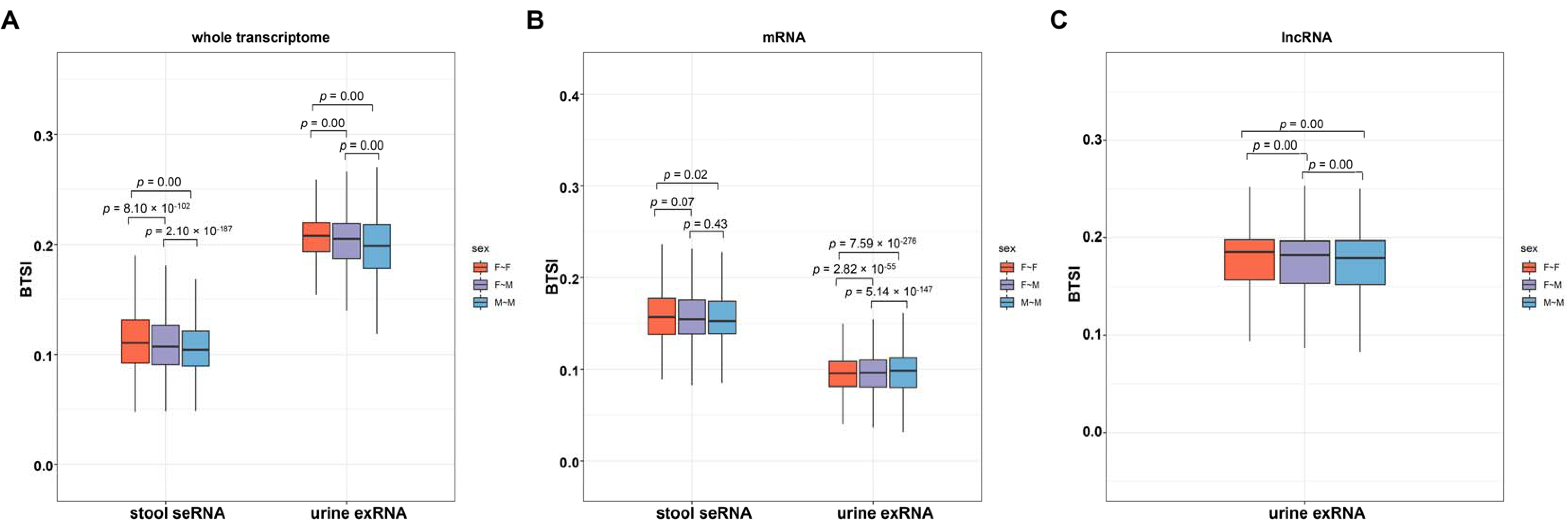
3.4. Various Factors Influence Biofluid–Tissue Correlation
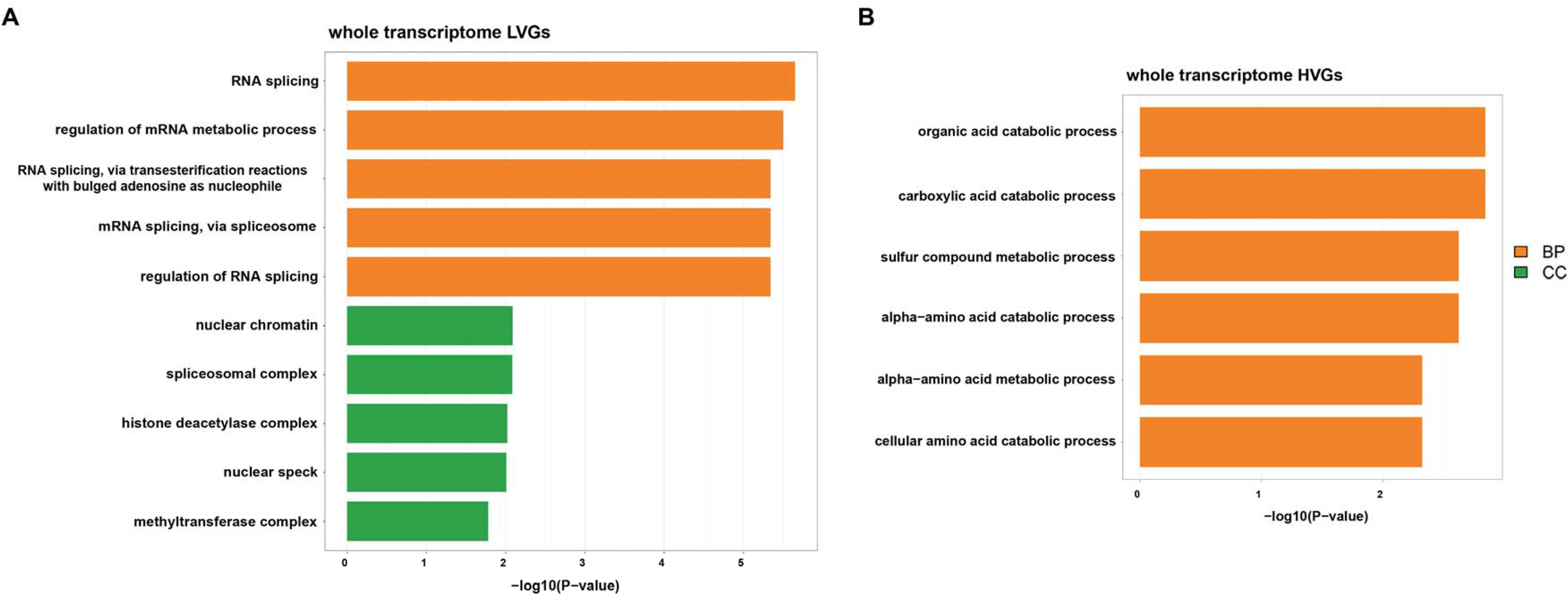
3.5. Function Played by Highly and Lowly Variable Component of Biofluid Transcriptome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Lanschot, M.C.J.; Bosch, L.J.W.; de Wit, M.; Carvalho, B.; Meijer, G.A. Early detection: The impact of genomics. Virchows Arch. 2017, 471, 165–173. [Google Scholar] [CrossRef]
- Proctor, G.B. The physiology of salivary secretion. Periodontology 2016, 70, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Duan, J.; Liu, T.; Smith, R.D.; Qian, W.J. Contributions of immunoaffinity chromatography to deep proteome profiling of human biofluids. J. Chromatogr. B 2016, 1021, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Yang, Y.; Guo, Z.; Shao, C.; Sun, H.; Zhang, Y.; Sun, Y.; Liu, Y.; Song, Y.; Zhang, L.; et al. A Comparative Proteomics Analysis of Five Body Fluids: Plasma, Urine, Cerebrospinal Fluid, Amniotic Fluid, and Saliva. Proteomics. Clin. Appl. 2018, 12, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Recent and potential developments of biofluid analyses in metabolomics. J. Proteom. 2012, 75, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; Wild, E.J. Biofluid Biomarkers in Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 329–396. [Google Scholar] [CrossRef] [PubMed]
- Marcuello, M.; Vymetalkova, V.; Neves, R.P.L.; Duran-Sanchon, S.; Vedeld, H.M.; Tham, E.; van Dalum, G.; Flügen, G.; Garcia-Barberan, V.; Fijneman, R.J.; et al. Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol. Asp. Med. 2019, 69, 107–122. [Google Scholar] [CrossRef]
- Agoston, D.V.; Shutes-David, A.; Peskind, E.R. Biofluid biomarkers of traumatic brain injury. Brain Inj. 2017, 31, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Cui, Q. The relationship of human tissue microRNAs with those from body fluids. Sci. Rep. 2020, 10, 5644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas Analysis Reveals Distinct Extracellular RNA Cargo Types and Their Carriers Present across Human Biofluids. Cell 2019, 177, 463–477.e415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Hunt, G.J.; Freytag, S.; Bahlo, M.; Gagnon-Bartsch, J.A. dtangle: Accurate and robust cell type deconvolution. Bioinformatics 2019, 35, 2093–2099. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Barnell, E.K.; Kang, Y.; Wurtzler, E.M.; Griffith, M.; Chaudhuri, A.A.; Griffith, O.L. Noninvasive Detection of High-Risk Adenomas Using Stool-Derived Eukaryotic RNA Sequences as Biomarkers. Gastroenterology 2019, 157, 884–887.e883. [Google Scholar] [CrossRef] [Green Version]
- Radon, T.P.; Massat, N.J.; Jones, R.; Alrawashdeh, W.; Dumartin, L.; Ennis, D.; Duffy, S.W.; Kocher, H.M.; Pereira, S.P.; Guarner posthumous, L.; et al. Identification of a Three-Biomarker Panel in Urine for Early Detection of Pancreatic Adenocarcinoma. Clin. Cancer Res. 2015, 21, 3512–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G.; et al. A Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer at Initial Biopsy. JAMA Oncol. 2016, 2, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu. Rev. Pathol. 2020, 15, 419–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi-Frias, D.; Vakar-Lopez, F.; Coleman, I.M.; Plymate, S.R.; Reed, M.J.; Nelson, P.S. The effects of aging on the molecular and cellular composition of the prostate microenvironment. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nature reviews. Genetics 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejos, C.A.; Marioni, J.C.; Richardson, S. BASiCS: Bayesian Analysis of Single-Cell Sequencing Data. PLoS Comput. Biol. 2015, 11, e1004333. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Cui, C.; Zhou, Y.; Cui, Q. Comprehensive Analysis of RNA Expression Correlations between Biofluids and Human Tissues. Genes 2021, 12, 935. https://doi.org/10.3390/genes12060935
Sun R, Cui C, Zhou Y, Cui Q. Comprehensive Analysis of RNA Expression Correlations between Biofluids and Human Tissues. Genes. 2021; 12(6):935. https://doi.org/10.3390/genes12060935
Chicago/Turabian StyleSun, Ruya, Chunmei Cui, Yuan Zhou, and Qinghua Cui. 2021. "Comprehensive Analysis of RNA Expression Correlations between Biofluids and Human Tissues" Genes 12, no. 6: 935. https://doi.org/10.3390/genes12060935
APA StyleSun, R., Cui, C., Zhou, Y., & Cui, Q. (2021). Comprehensive Analysis of RNA Expression Correlations between Biofluids and Human Tissues. Genes, 12(6), 935. https://doi.org/10.3390/genes12060935