
A Methodological Framework to Discover Pharmacogenomic Interactions Based on Random Forests

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
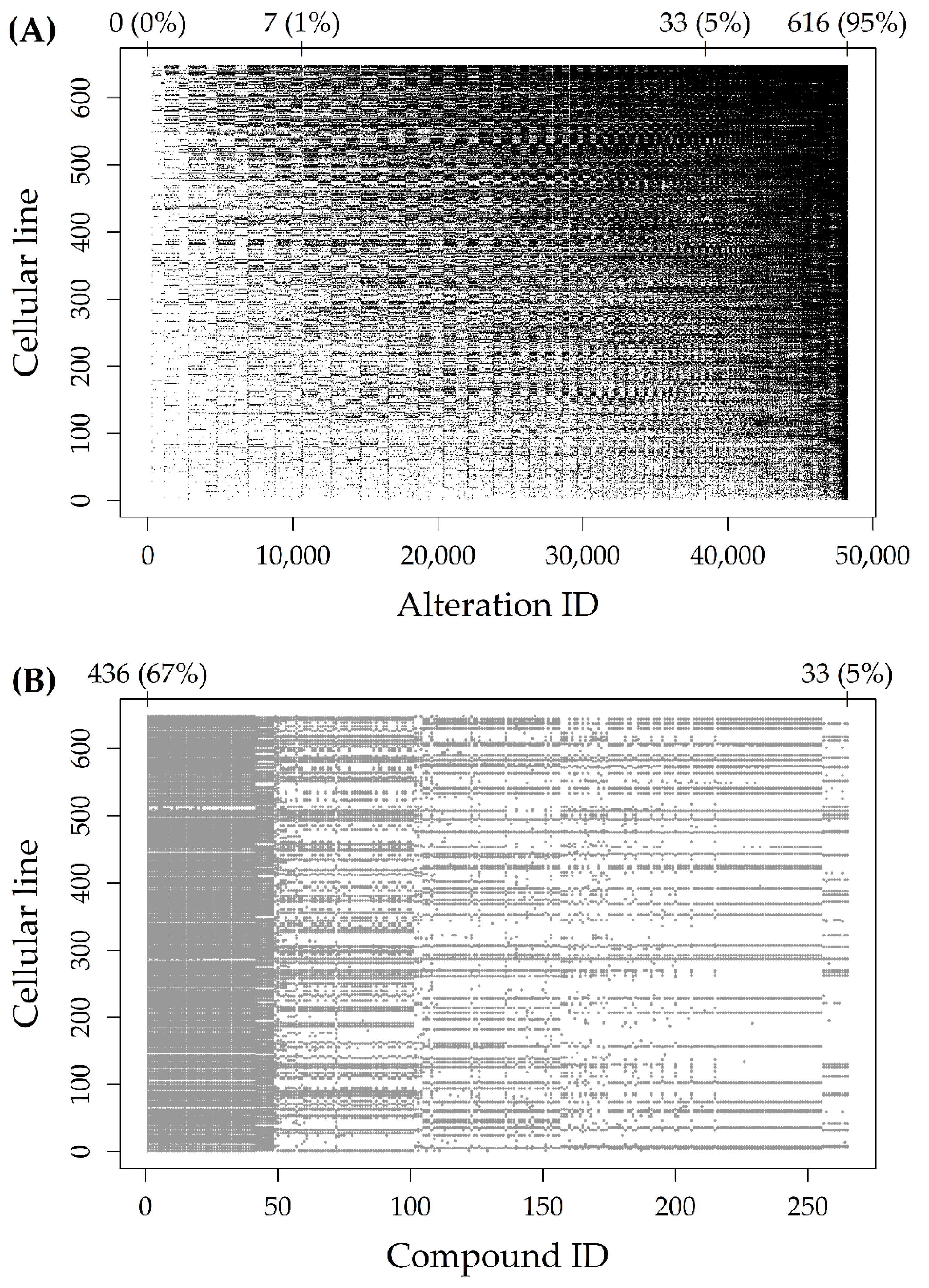
2.1. Alteration and Response Datasets
2.2. Random Forests
2.3. Data Reduction
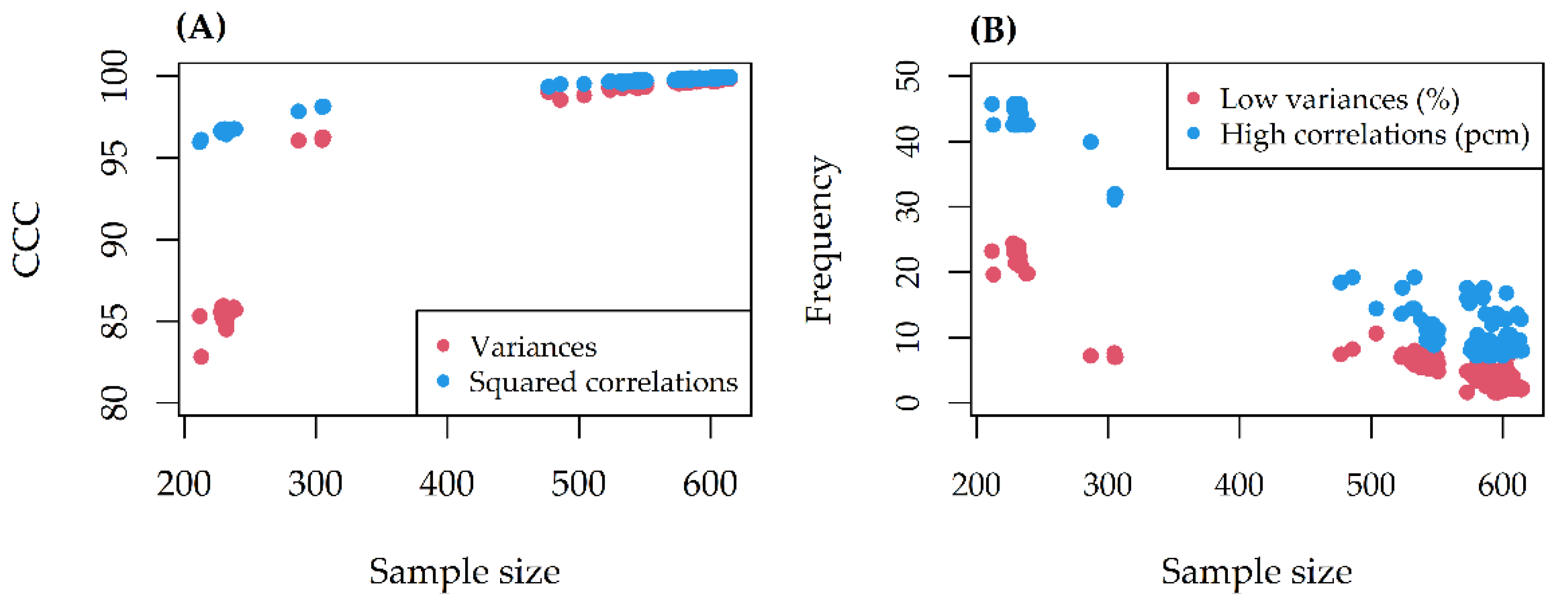
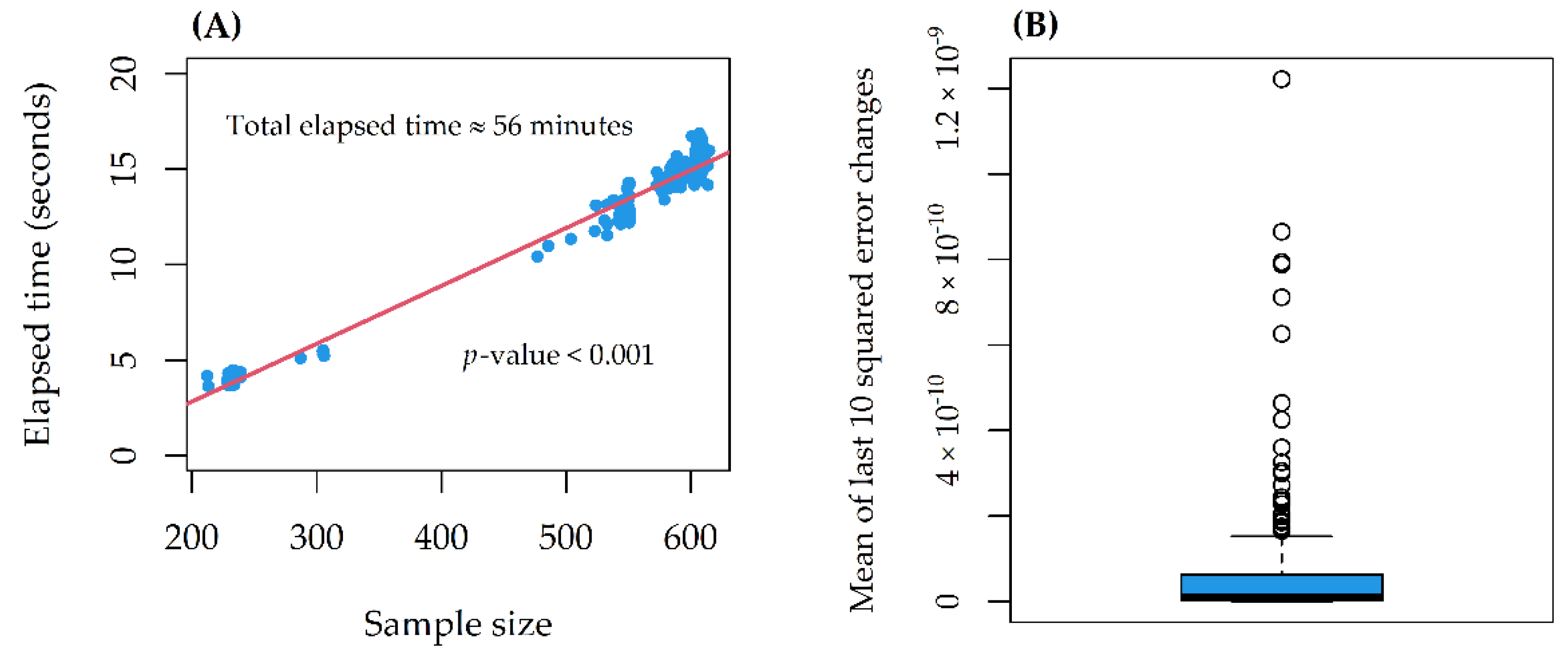
2.4. Predictive Performance
2.5. Variable Importance
2.6. Missing Values
2.7. Reporting Results
2.8. Discovering Drug-Gene Interactions
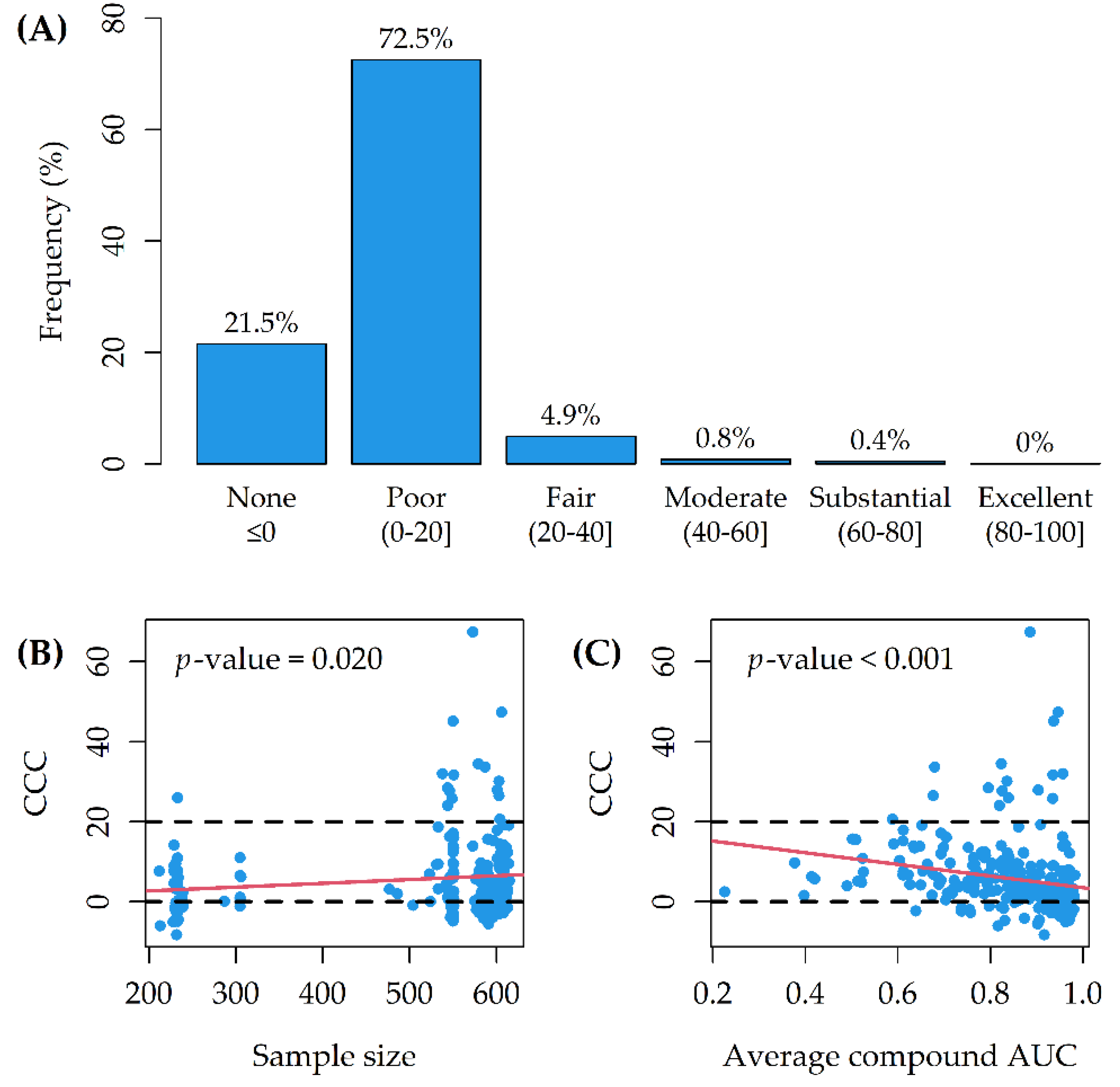
3. Results
4. Discussion
4.1. Strengths
4.2. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smida, M.; Nijman, S.M. Functional Drug–Gene Interactions in Lung Cancer. Expert Rev. Mol. Diagn. 2012, 12, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, K.K.; Duijf, P.H. Complexities of Pharmacogenomic Interactions in Cancer. Mol. Cell. Oncol. 2020, 7, 1735910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.; Havener, T.; Everitt, L.; McLeod, H.; Motsinger-Reif, A. A Comparison of Association Methods for Cytotoxicity Mapping in Pharmacogenomics. Front. Genet. 2011, 2, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Pang, L. Comparing Statistical Methods for Quantifying Drug Sensitivity Based on in vitro Dose–Response Assays. Assay Drug Dev. Technol. 2012, 10, 88–96. [Google Scholar] [CrossRef]
- Cokelaer, T.; Chen, E.; Iorio, F.; Menden, M.P.; Lightfoot, H.; Saez-Rodriguez, J.; Garnett, M.J. GDSCTools for Mining Pharmacogenomic Interactions in Cancer. Bioinformatics 2018, 34, 1226–1228. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Aittokallio, T. Machine Learning and Feature Selection for Drug Response Prediction in Precision Oncology Applications. Biophys. Rev. 2019, 11, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.Q.; Chen, L.; Cooper, G.F.; Young, J.D.; Lu, X. Precision Oncology beyond Targeted Therapy: Combining Omics Data with Machine Learning Matches the Majority of Cancer Cells to Effective Therapeutics. Mol. Cancer Res. 2018, 16, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, I.S.; Neto, E.C.; Guinney, J.; Friend, S.H.; Margolin, A.A. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. In Biocomputing 2014; World Scientific: Singapore, 2013; pp. 63–74. [Google Scholar]
- Huang, J.; Chen, J.; Zhang, B.; Zhu, L.; Cai, H. Evaluation of Gene-Drug Common Module Identification Methods Using Pharmacogenomics Data. Brief. Bioinform. 2021, 22, bbaa087. [Google Scholar] [CrossRef]
- Cramer, D.; Mazur, J.; Espinosa, O.; Schlesner, M.; Hübschmann, D.; Eils, R.; Staub, E. Genetic Interactions and Tissue Specificity Modulate the Association of Mutations with Drug Response. Mol. Cancer Ther. 2020, 19, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X.; Liu, X.S. Predicting Anticancer Drug Responses Using a Dual-Layer Integrated Cell Line-Drug Network Model. PLoS Comput. Biol. 2015, 11, e1004498. [Google Scholar] [CrossRef]
- Ammad-Ud-Din, M.; Khan, S.A.; Wennerberg, K.; Aittokallio, T. Systematic Identification of Feature Combinations for Predicting Drug Response with Bayesian Multi-View Multi-Task Linear Regression. Bioinformatics 2017, 33, i359–i368. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Riddick, G.; Song, H.; Ahn, S.; Walling, J.; Borges-Rivera, D.; Zhang, W.; Fine, H.A. Predicting in vitro Drug Sensitivity Using Random Forests. Bioinformatics 2011, 27, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and Regression by RandomForest. R News 2002, 2, 18–22. [Google Scholar]
- Han, S.; Kim, H. On the Optimal Size of Candidate Feature Set in Random Forest. Appl. Sci. 2019, 9, 898. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Genuer, R.; Poggi, J.-M.; Tuleau-Malot, C. Variable Selection Using Random Forests. Pattern Recognit. Lett. 2010, 31, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Chavent, M.; Genuer, R.; Saracco, J. Combining Clustering of Variables and Feature Selection Using Random Forests. Commun. Stat. Simul. Comput. 2021, 50, 426–445. [Google Scholar] [CrossRef] [Green Version]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning; Springer: New York, NY, USA, 2013. [Google Scholar]
- Lawrence, I.; Lin, K. A Concordance Correlation Coefficient to Evaluate Reproducibility. Biometrics 1989, 255–268. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The Measurement of Observer Agreement for Categorical Data. Biometrics 1977, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Strobl, C.; Boulesteix, A.-L.; Zeileis, A.; Hothorn, T. Bias in Random Forest Variable Importance Measures: Illustrations, Sources and a Solution. BMC Bioinform. 2007, 8, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Lunetta, K.L.; Hayward, L.B.; Segal, J.; Van Eerdewegh, P. Screening Large-Scale Association Study Data: Exploiting Interactions Using Random Forests. BMC Genet. 2004, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.J.; Berger, J.O.; Johannesson, M.; Nosek, B.A.; Wagenmakers, E.-J.; Berk, R.; Bollen, K.A.; Brembs, B.; Brown, L.; Camerer, C.; et al. Redefine Statistical Significance. Nat. Hum. Behav. 2018, 2, 6–10. [Google Scholar] [CrossRef]
- Nguyen, L.; Dang, C.C.; Ballester, P.J. Systematic Assessment of Multi-Gene Predictors of Pan-Cancer Cell Line Sensitivity to Drugs Exploiting Gene Expression Data. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hensman, J.; Kutkaite, G.; Toh, T.S.; Galhoz, A.; Dry, J.R.; Saez-Rodriguez, J.; Garnett, M.J.; Menden, M.P.; Dondelinger, F.; et al. A Statistical Framework for Assessing Pharmacological Responses and Biomarkers Using Uncertainty Estimates. Elife 2020, 9, e60352. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Pal, R. Analyzing Drug Sensitivity Prediction Based on Dose Response Curve Characteristics. In Proceedings of the 2016 IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), Las Vegas, NV, USA, 24–27 February 2016; pp. 140–143. [Google Scholar]
- Lu, H.; Villafane, N.; Dogruluk, T.; Grzeskowiak, C.L.; Kong, K.; Tsang, Y.H.; Zagorodna, O.; Pantazi, A.; Yang, L.; Neill, N.J.; et al. Engineering and Functional Characterization of Fusion Genes Identifies Novel Oncogenic Drivers of Cancer. Cancer Res. 2017, 77, 3502–3512. [Google Scholar] [CrossRef] [Green Version]
- Karan, G.; Wang, H.; Chakrabarti, A.; Karan, S.; Liu, Z.; Xia, Z.; Gundluru, M.; Moreton, S.; Saunthararajah, Y.; Jackson, M.W.; et al. Identification of a Small Molecule That Overcomes HdmX-Mediated Suppression of P53. Mol. Cancer Ther. 2016, 15, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Curioni-Fontecedro, A.; Nechushtan, H.; Shih, J.-Y.; Liao, W.-Y.; Gautschi, O.; Spataro, V.; Unk, M.; Yang, J.C.-H.; Lorence, R.M.; et al. Activity of Afatinib in Heavily Pretreated Patients with ERBB2 Mutation–Positive Advanced NSCLC: Findings from a Global Named Patient Use Program. J. Thorac. Oncol. 2018, 13, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.V.; Hao, X.; Aman, A.; Luchman, H.A.; Weiss, S. EGFR Blockade in GBM Brain Tumor Stem Cells Synergizes with JAK2/STAT3 Pathway Inhibition to Abrogate Compensatory Mechanisms in vitro and in Vivo. Neuro-Oncol. Adv. 2020, 2, vdaa020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with Open Crowdsource Efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Dang, C.C.; Peón, A.; Ballester, P.J. Unearthing New Genomic Markers of Drug Response by Improved Measurement of Discriminative Power. BMC Med. Genom. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Bronte, G.; Rizzo, S.; La Paglia, L.; Adamo, V.; Siragusa, S.; Ficorella, C.; Santini, D.; Bazan, V.; Colucci, G.; Gebbia, N.; et al. Driver Mutations and Differential Sensitivity to Targeted Therapies: A New Approach to the Treatment of Lung Adenocarcinoma. Cancer Treat. Rev. 2010, 36, S21–S29. [Google Scholar] [CrossRef]
- Zsákai, L.; Sipos, A.; Dobos, J.; Erős, D.; Szántai-Kis, C.; Bánhegyi, P.; Pató, J.; Őrfi, L.; Matula, Z.; Mikala, G.; et al. Targeted Drug Combination Therapy Design Based on Driver Genes. Oncotarget 2019, 10, 5255. [Google Scholar] [CrossRef] [Green Version]
- Scholl, C.; Fröhling, S. Exploiting Rare Driver Mutations for Precision Cancer Medicine. Curr. Opin. Genet. Dev. 2019, 54, 1–6. [Google Scholar] [CrossRef]
- Liggett, L.A.; Sharma, A.; De, S.; DeGregori, J. FERMI: A Novel Method for Sensitive Detection of Rare Mutations in Somatic Tissue. G3 Genes Genomes Genet. 2019, 9, 2977–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum: Hillsdale, NJ, USA, 1988. [Google Scholar]
- The Cancer Cell Line Encyclopedia and Genomics of Drug sensitivity in Cancer investigators. Pharmacogenomic Agreement between Two Cancer Cell Line Data Sets. Nature 2015, 528, 84. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, P.; Safikhani, Z.; El-Hachem, N.; Wang, D.; She, A.; Olsen, C.; Freeman, M.; Selby, H.; Gendoo, D.M.; Grossmann, P.; et al. PharmacoGx: An R Package for Analysis of Large Pharmacogenomic Datasets. Bioinformatics 2016, 32, 1244–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, R.; Matlock, K.; Ghosh, S.; Pal, R. Heterogeneity Aware Random Forest for Drug Sensitivity Prediction. Sci. Rep. 2017, 7, 11347. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | No. (%) |
|---|---|
| Lung | 134 (20.7%) |
| Hematopoietic and lymphoid tissue | 111 (17.1%) |
| Breast | 46 (7.1%) |
| Large intestine | 43 (6.6%) |
| Central nervous system | 39 (6%) |
| Skin | 36 (5.6%) |
| Ovary | 31 (4.8%) |
| Pancreas | 28 (4.3%) |
| Esophagus | 24 (3.7%) |
| Stomach | 22 (3.4%) |
| Liver | 17 (2.6%) |
| Urinary tract | 17 (2.6%) |
| Upper aero digestive tract | 16 (2.5%) |
| Soft tissue | 15 (2.3%) |
| Kidney | 14 (2.2%) |
| Autonomic ganglia | 12 (1.9%) |
| Bone | 11 (1.7%) |
| Endometrium | 10 (1.5%) |
| Thyroid | 9 (1.4%) |
| Pleura | 6 (0.9%) |
| Prostate | 5 (0.8%) |
| Biliary tract | 1 (0.2%) |
| Small intestine | 1 (0.2%) |
| Compound | CCC (95% CI) | Mean AUC | Sample Size | Number of Influential Alterations | Alteration 1 | Alteration 2 |
|---|---|---|---|---|---|---|
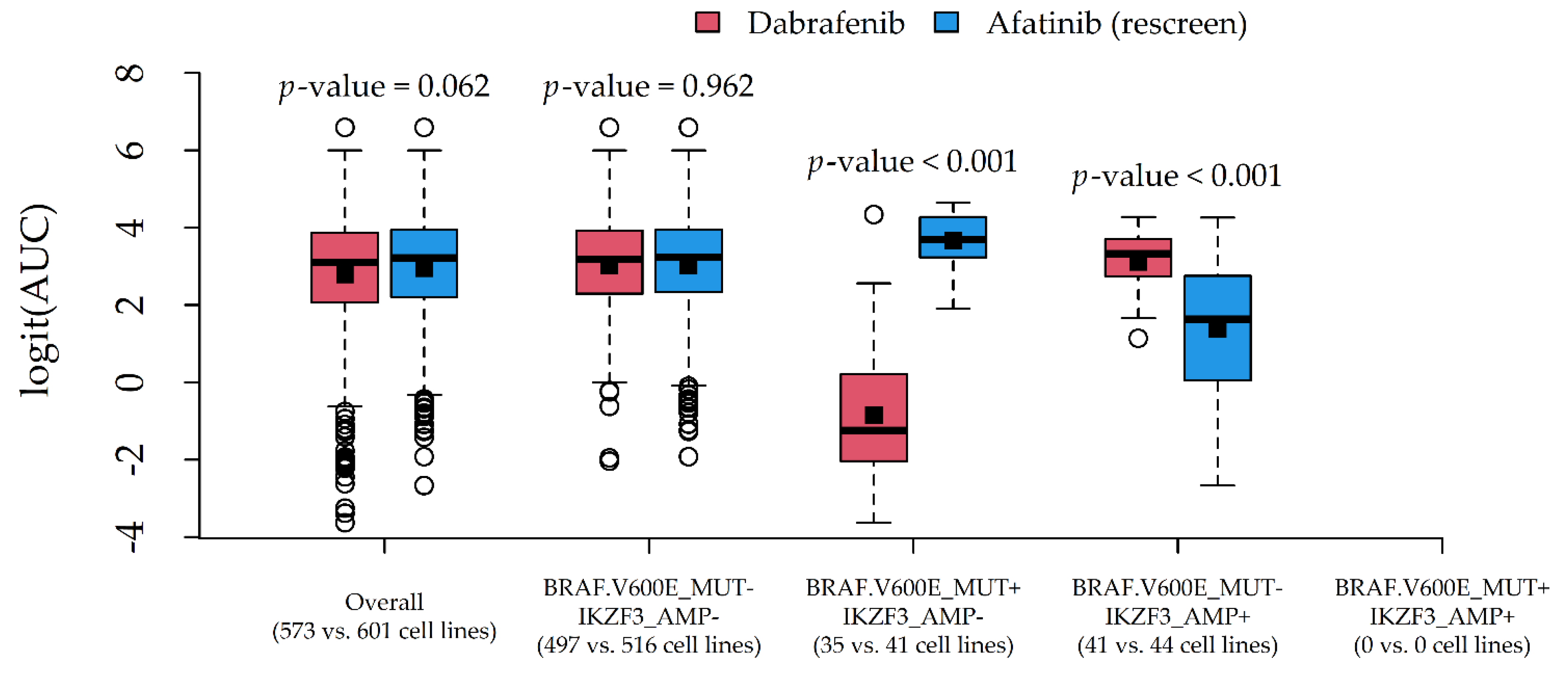
| Dabrafenib | 67.4 (63.1, 71.3) | 0.886 | 573 | 4 | BRAF.V600E_MUT | BRAF_MUT |
| PLX4720 (rescreen) | 47.3 (42.3, 52.1) | 0.946 | 606 | 5 | BRAF.V600E_MUT | BRAF_MUT |
| PLX4720 | 45.1 (39.7, 50.2) | 0.937 | 550 | 8 | BRAF.V600E_MUT | BRAF_MUT |
| RDEA119 (rescreen) | 34.5 (29.0, 39.7) | 0.824 | 579 | 9 | BRAF.V600E_MUT | KRAS_MUT |
| Trametinib | 33.6 (27.9, 39.0) | 0.680 | 587 | 16 | BRAF.V600E_MUT | KRAS_MUT |
| SB590885 | 32.0 (26.2, 37.5) | 0.957 | 538 | 3 | BRAF.V600E_MUT | BRAF_MUT |
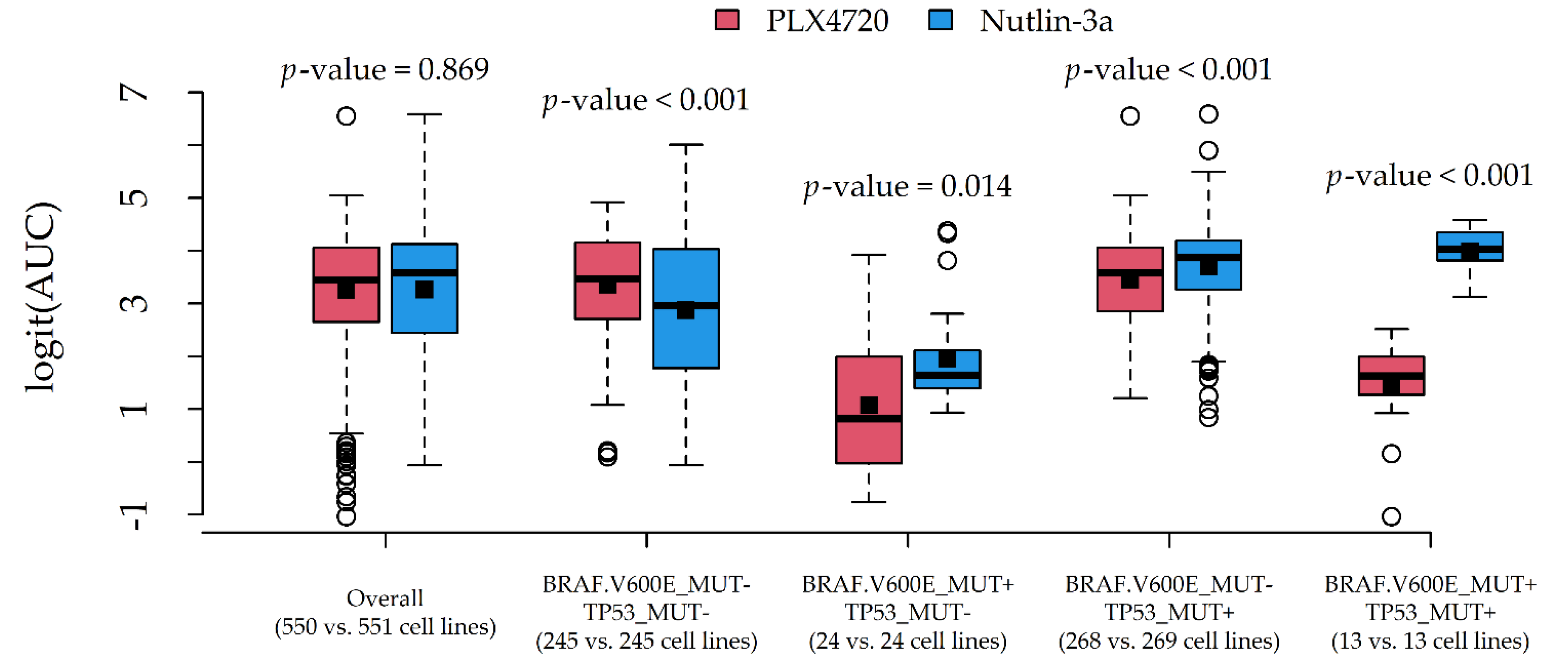
| Nutlin-3a | 31.7 (25.7, 37.4) | 0.936 | 551 | 10 | TP53_MUT | MAP2K4_DEL |
| AZD6244 | 30.1 (24.4, 35.5) | 0.836 | 603 | 6 | BRAF.V600E_MUT | KRAS_MUT |
| RDEA119 | 28.4 (22.6, 34.0) | 0.796 | 544 | 7 | BRAF.V600E_MUT | KRAS_MUT |
| Afatinib (rescreen) | 27.9 (22.0, 33.6) | 0.904 | 601 | 6 | IKZF3_AMP | ERBB2_AMP |
| PD-0325901 | 27.6 (21.7, 33.3) | 0.826 | 546 | 8 | BRAF.V600E_MUT | KRAS_MUT |
| (5Z)-7-Oxozeaenol | 26.5 (21.6, 31.2) | 0.677 | 603 | 9 | BRAF.V600E_MUT | NRAS_MUT |
| Alteration | Significance Frequency | Cluster ID | Cluster Size | Compound 1 | Compound 2 |
|---|---|---|---|---|---|
| BRAF.V600E_MUT | 11 | 184 | 1 | Dabrafenib | PLX4720 (rescreen) |
| BRAF_MUT | 10 | 385 | 1 | Dabrafenib | PLX4720 (rescreen) |
| NRAS_MUT | 6 | 185 | 1 | RDEA119 (rescreen) | Trametinib |
| KRAS.G12_13_MUT | 5 | 431 | 1 | RDEA119 (rescreen) | Trametinib |
| KRAS_MUT | 5 | 464 | 1 | RDEA119 (rescreen) | Trametinib |
| CREBBP_MUT | 4 | 384 | 1 | Dabrafenib | Trametinib |
| FHL5_DEL | 4 | 116 | 1 | PLX4720 (rescreen) | PLX4720 |
| BCL9_AMP | 3 | 358 | 1 | RDEA119 (rescreen) | Trametinib |
| ARHGAP40_AMP | 2 | 303 | 1 | Trametinib | PD-0325901 |
| CCDC66_DEL | 2 | 383 | 1 | Dabrafenib | Nutlin-3a |
| MAP2K4_DEL | 2 | 437 | 1 | RDEA119 (rescreen) | Nutlin-3a |
| RAF1_DEL | 2 | 225 | 1 | Trametinib | Afatinib (rescreen) |
| TP53_MUT | 2 | 501 | 1 | Nutlin-3a | (5Z)-7-Oxozeaenol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasola, S.; Cilluffo, G.; Montalbano, L.; Malizia, V.; Ferrante, G.; La Grutta, S. A Methodological Framework to Discover Pharmacogenomic Interactions Based on Random Forests. Genes 2021, 12, 933. https://doi.org/10.3390/genes12060933
Fasola S, Cilluffo G, Montalbano L, Malizia V, Ferrante G, La Grutta S. A Methodological Framework to Discover Pharmacogenomic Interactions Based on Random Forests. Genes. 2021; 12(6):933. https://doi.org/10.3390/genes12060933
Chicago/Turabian StyleFasola, Salvatore, Giovanna Cilluffo, Laura Montalbano, Velia Malizia, Giuliana Ferrante, and Stefania La Grutta. 2021. "A Methodological Framework to Discover Pharmacogenomic Interactions Based on Random Forests" Genes 12, no. 6: 933. https://doi.org/10.3390/genes12060933
APA StyleFasola, S., Cilluffo, G., Montalbano, L., Malizia, V., Ferrante, G., & La Grutta, S. (2021). A Methodological Framework to Discover Pharmacogenomic Interactions Based on Random Forests. Genes, 12(6), 933. https://doi.org/10.3390/genes12060933