
Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain

Abstract
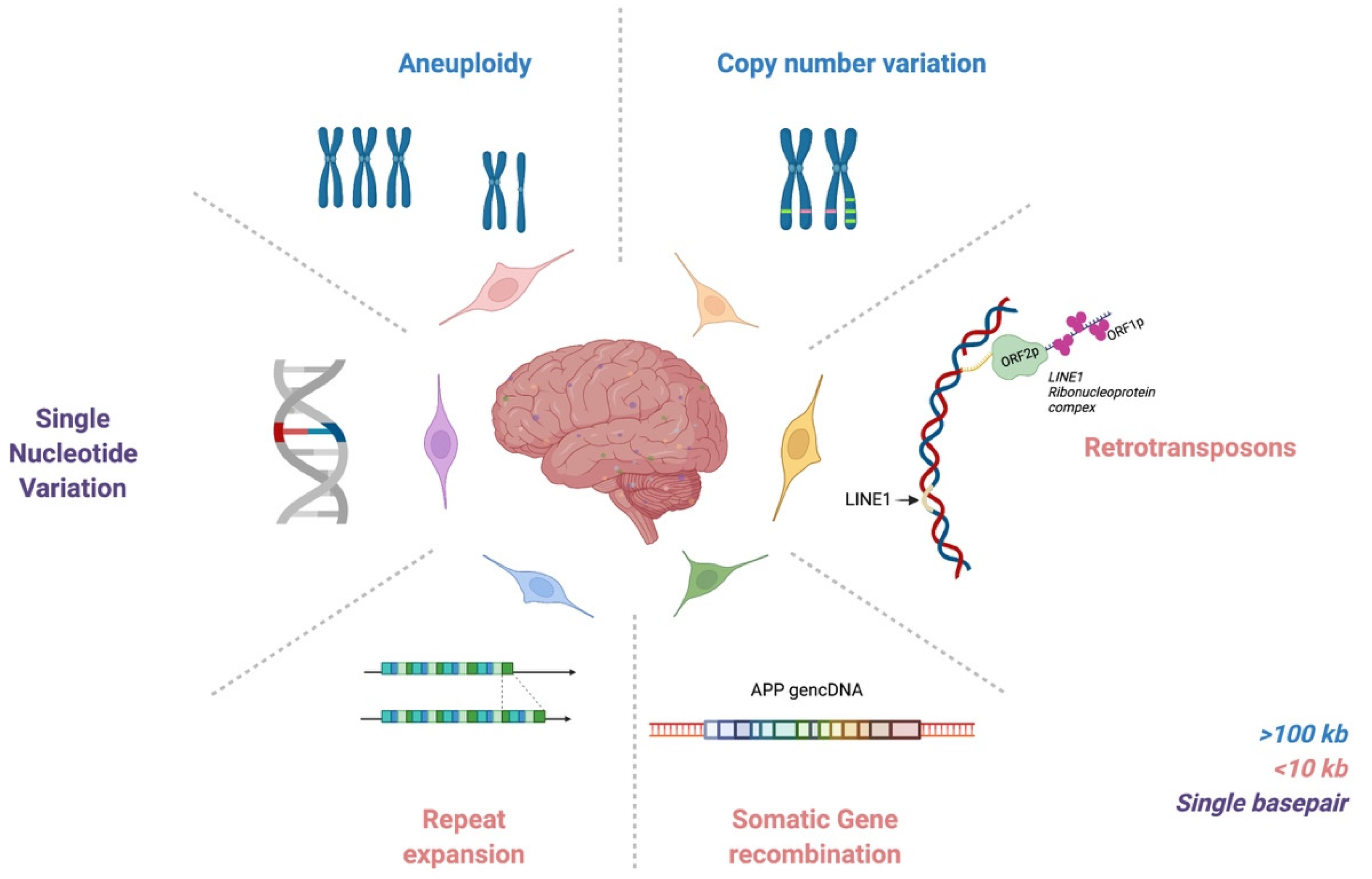
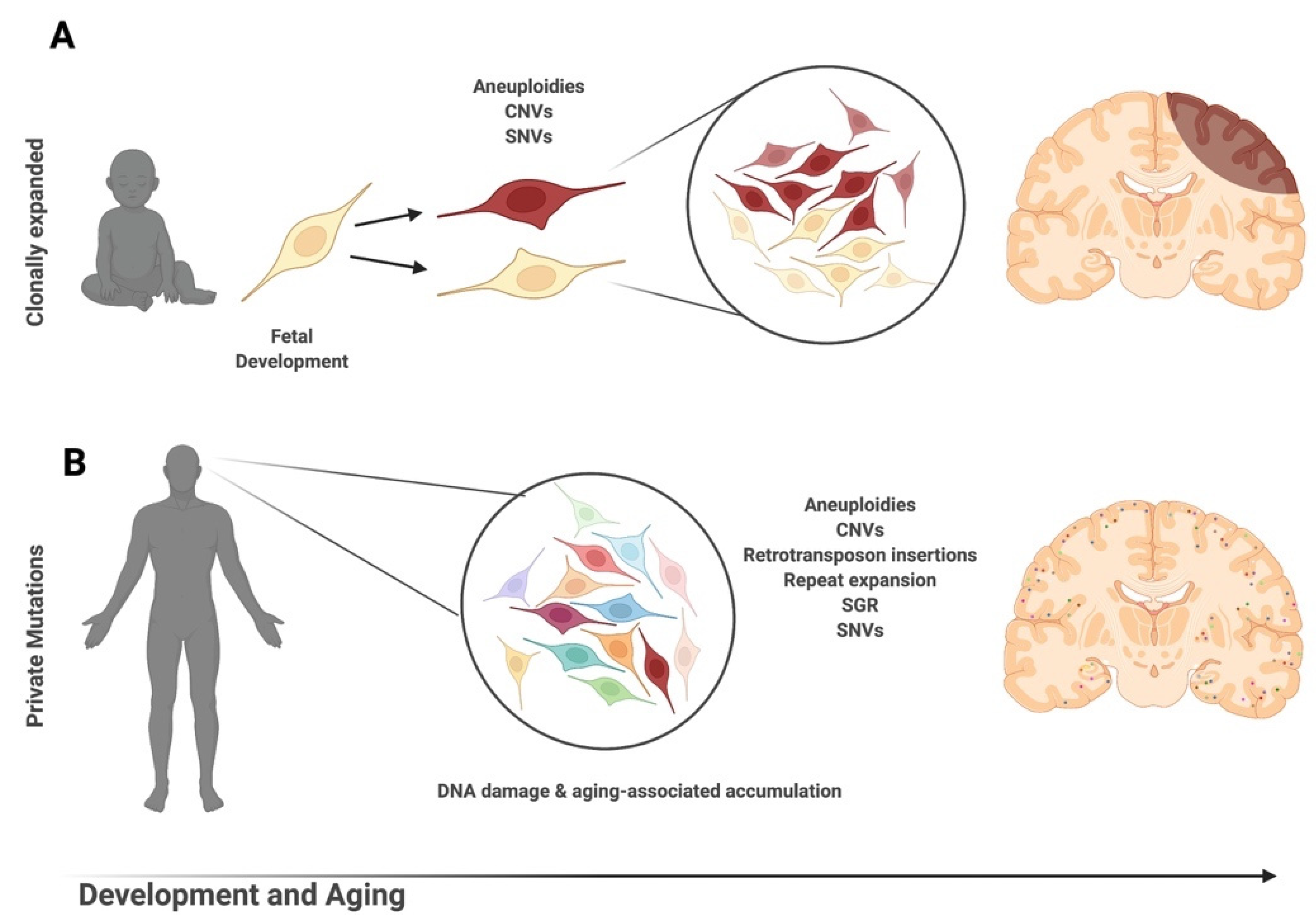
:1. Introduction
2. DNA Content Variation & Aneuploidy
2.1. DNA Content Variation & Aneuploidy in Health and Aging
2.2. DNA Content Variation & Aneuploidy in Neurodegenerative Disease
3. Copy Number Variation
3.1. Somatic Copy Number Variation in Health & Aging
3.2. Somatic Copy Number Variation in Neurodegenerative Disease
4. Retrotransposons
4.1. Retrotransposons in the Normal Brain
4.2. Somatic Retrotransposition in Neurodegenerative Disease
4.3. Aging-Associated Neurological Degeneration & Retrotransposon Copy Number/Expression
5. Somatic Repeat Expansion
6. Somatic Gene Recombination
7. Somatic Single Nucleotide Variations
7.1. Somatic Single Nucleotide Variations in Development & Aging
7.2. Somatic Single Nucleotide Variation in Neurodegenerative Disease
8. Future Technologies, Research, & Therapeutics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
| Disease | Somatic Variation | DNA Source | Technique | Somatic Finding | Reference |
|---|---|---|---|---|---|
| AD | DCV | 16 SAD (7M:9F) & 16 ND (7M:9F); mean age 75 yo. 5 postmortem cortical regions | Slide-based cytometry | ↑ DC in all AD cortical regions | Arendt et al., 2015 [45] |
| CNV | 32 SAD (14M:18F, 62–101 yo.) & 40 ND (15M:25F, 17–103 yo.) postmortem FR, CBLM | qPCR; PNA-FISH; flow cytometry | ↑ DC & ↑APP copy number in SAD | Bushman et al., 2015 [31] | |
| 20 SAD, 20 PD/LBD, & 14 ND; mean age 80–84 yo. 5 postmortem brain regions | Whole exome sequencing | 1 APP gain in AD case | Keogh et al., 2018 [66] | ||
| 72 SAD (61–105 yo.) & 58 non-AD (18–97 yo.) postmortem ERC | Gene enrichment & amplicon sequencing | No CNVs in APP, PSEN1, PSEN2, MAPT | Sala Frigerio et al., 2015 [67] | ||
| RTsp | 422 SAD & 201 ND postmortem FR | RNA-seq | ↑ LINE1 & HERVk expression | Guo et al., 2018 [128] | |
| SGR | 7 SAD (1M:6F, 72–88 yo.) & 6 ND (3M:3F, 80–94 yo.) postmortem FR, CBLM | Amplicon sequencing; APP exonic pulldown; DISH | ↑ APP gencDNAs in AD NeuN+ FR | Lee et al., 2018 [156] | |
| 52 SAD (16M:36F, 70–96 yo.) & 11 ND (7M:4F, 57–89 yo.) paired blood & postmortem laser-captured HIF | Whole exome sequencing | APP IEJs in AD HPC | Park et al., 2019 [160] | ||
| SNVs | 2 related individuals (58 yo. F, 39 yo. F) blood & postmortem CTX | Allele-specific oligonucleotide hybridization | Mosaic PSEN1 SNV in mother; germline heterozygous PSEN1 SNV in child | Beck et al., 2004 [174] | |
| 17 SAD (5M:12F), 2 VD (1M:1F), & 2 ND (2M) 46–94 yo. paired blood & postmortem HPC, CBLM | Whole exome sequencing | AD brain-specific SNVs in AD assoc. genes | Parcerisas et al., 2014 [176] | ||
| 72 SAD (61–105 yo.) & 58 non-AD (18–97 yo.) postmortem ERC | Gene enrichment & amplicon sequencing | 2 MAPT & 1 PSEN2 SNVs in AD & non-AD | Sala Frigerio et al., 2015 [67] | ||
| 372 EOAD (>66 years old at diagnosis), 73 LOAD, 1 FAD, & 52 ND blood & postmortem brain | smMIP assay & amplicon sequencing | 9 candidate SNVs of benign/unknown sig. | Nicolas et al., 2018 [182] | ||
| 4 EOAD (59–68 yo.), 4 LOAD (79–89), 8 ND (53–88) blood & postmortem TC | Whole exome sequencing | 1 brain-specific SNV in CD55 in LOAD | Helgadottir et al., 2019 [183] | ||
| 20 SAD & 20 ND postmortem OB, HPC & 4 cortical regions | ddPCR; RNA-seq | Autism-assoc. ADNP SNVs in AD; 104 genes with disease-causing SNVs in AD | Ivashko-Pachima et al., 2019 [185] | ||
| 52 SAD (16M:36F, 70–96 yo.) & 11 ND (7M:4F, 57–89 yo.) paired blood & postmortem laser-captured HIF | Whole exome sequencing | 1 PIN1 pathogenic mutation in SAD | Park et al., 2019 [160] | ||
| ALS | CNV | 32 SALS (22M:10F; 47–84 yo.) & ND (18M:6F) blood & postmortem brain | Microarray | 24 CNVs in genic/promoter regions | Pamphlett et al., 2011 [69] |
| RTsp | 25 SALS, 3 FALS (mean 63 yo.) & 12 ND (mean 60 yo.) 4 CTX regions | RTqPCR | ↑ HERVk pol expression in ALS | Douville et al., 2010 [125] | |
| 148 SALS, 11 other neurologic disease, & 17 ND postmortem CTX | RNA-seq | ↑ LINE1 expression in ALS | Tam et al., 2019 [123] | ||
| RE | 19 SALS (9M:7F:3unknown; 50–79 yo.), C9ORF72 expansion negative; postmortem spinal cord | RepeatPrimer PCR & amplicon size genotyping | No somatic expansion of C9ORF72 repeat | Ross et al., 2019 [155] | |
| ALS with or without C9ORF72 repeat expansion blood, CNS, non-neural tissues | Southern blot | Intra-individual variation of C9ORF72 repeat | Buchman et al., 2013 [152], Dols-Icardo et al., 2014 [153], Nordin et al., 2015 [154] | ||
| SNV | 2 related individuals (33 yo. M, 50 yo. F, living) blood & saliva | Whole exome sequencing | FUS mosaic SNV in mother; germline heterozygous FUS SNV in child | Hisahara et al., 2021 [175] | |
| A-T | RTsp | 7 A-T & 7 ND, 8–28 yo. laser capture of postmortem HPC | Taqman-based qPCR for ORF2 sequence | ↑ LINE1 copy number in A-T | Coufal et al., 2011 [112] |
| 4 A-T, 2 RT, 72 other, & 20 ND postmortem neural & non-neural tissue | Whole-genome sequencing | ↑ LINE1 copy number in A-T cortex | Jacob-Hirsch et al., 2018 [113] | ||
| FTLD | RTsp | FTLD & ND brain | Crosslinking-immunoprecipitation sequencing | ↓ binding of RTsp by TDP-43 in FTLD brains | Li et al., 2012 [120] |
| RE | FTLD with or without C9ORF72 repeat expansion blood, CNS, non-neural tissues | Southern blot | Intra-individual variation of C9ORF72 repeat | Buchman et al., 2013 [152], Dols-Icardo et al., 2014 [153], Nordin et al., 2015 [154] | |
| HD | RE | 3 HD (27–40 yo.) postmortem striatum | PCR amplification; small-pool PCR | ↑ CAG repeats in striatal cells | Kennedy et al., 2003 [133] |
| 5 HD (3M:2F, 40–64 yo.) postmortem striatum & TC | PCR amplification & Southern blot | ↑ CAG repeats in neurons vs. glia | Shelbourne et al., 2007 [136] | ||
| 24 HD young onset (20–41yo.) & 24 old onset (40–81 yo.) postmortem CTX & CBLM | Small-pool PCR | ↑ repeat size assoc. with young onset | Swami et al., 2009 [134] | ||
| 7 adult-onset HD (2M:5F, 39–66 yo.) & 1 juvenile- onset HD (1M, 6 yo.), postmortem CNS & PNS tissue | Repeat length genotyping | ↑ ATXN1 CAG repeats in brain tissues | Mouro Pinto et al., 2020 [148] | ||
| MSA | CNV | 5 MSA (55–76 yo.) & 30 ND (59–94 yo.) postmortem SN dopaminergic neurons | FISH | ↑ SNCA copy number in MSA | Mokretar et al., 2018 [63] |
| 18 MSA (5M:13F, 52–82 yo.) & 17 ND (10M:7F, 59–92 yo.), postmortem cingulate CTX, CBLM | FISH; whole-genome sequencing | ↑ SNCA copy number in MSA | Perez-Rodriguez et al., 2019 [64] | ||
| PD | CNV | 8 PD (5M:3F, 63–81yo.) & 26 ND (18M:8F, 44–85yo.) postmortem FR | Microarray | CNVs detected in PD candidate genes (not SNCA) | Pamphlett et al., 2012 [54] |
| 2 PD living donors (40 yo. M, 23yo. M): mucosal cells, blood | FISH | ↑ in 4p22.1 locus of SNCA in PD mucosa | Perandones et al., 2014 [62] | ||
| 41 PD (56–83 yo.) & 30 ND (59–92 yo.) postmortem SN dopaminergic neurons | FISH | ↑ SNCA copy number in PD | Mokretar et al., 2018 [63] | ||
| 26 PD (20M:6F, 60–83 y.o) & 18 ND (10M:7F, 59–92 yo.) cingulate CTX & CBLM | FISH; whole-genome sequencing | ↑ SNCA copy number in PD | Perez-Rodriguez et al., 2019 [64] | ||
| SNV | 28 PD (17M:11F, 62–90 yo.) postmortem SN & CBLM | HRM analysis of amplicons | No SNVs within SNCA | Proukakis et al., 2013 [178] | |
| 511 idiopathic PD (age of onset 61 yo.) postmortem CBLM, SN, FR | HRM analysis of amplicons | No SNVs within SNCA | Proukakis et al., 2014 [179] | ||
| 20 PD/LBD (67–91 yo.) & 15 ND (64–97) blood & 4 postmortem brain regions | Gene enrichment panels | Brain-specific SNVs in neurodegenerative genes | Keogh et al., 2018 [180] | ||
| 25 sporadic PD (21M:4F, 55–88 yo.), 1 familial PD, & 12 ND (4M:8F, 69–104 yo.); postmortem SN | Gene enrichment panel; ddPCR | No disease-relevant SNVs detected | Leija-Salazar et al., 2020 [177] | ||
| RTT | RTsp | 4 A-T, 2 RT, 72 other, & 20 ND postmortem neural & non-neural tissue | Whole-genome sequencing | ↑ LINE1 copy number in RTT | Jacob-Hirsch et al., 2018 [113] |
| 5 RTT (5F, 17–21 yo.) & 5 ND (5F, 16–25 yo.) postmortem CNS & peripheral tissue | PCR-based targeted bulk sequencing | ↑ LINE1 insertions in CTX neurons | Zhao et al., 2019 [115] | ||
| SCA1 | RE | 1 SCA1 postmortem CNS & peripheral tissue | Repeat length genotyping | ↑ ATXN1 CAG repeats in brain tissues | Mouro Pinto et al., 2020 [148] |
References
- Cajal, S.R.y. Studies on the Cerebral Cortex; University of Madrid: Madrid, Spain, 1901; Volume 1. [Google Scholar]
- Cajal, S.R.y. Histologie du Système Nerveux de L’homme et des Vertébrés; Maloine: Paris, French, 1909; ISBN 9782011774125. [Google Scholar]
- de Castro, F.; López-Mascaraque, L.; De Carlos, J.A. Cajal: Lessons on brain development. Brain Res. Rev. 2007, 55, 481–489. [Google Scholar] [CrossRef]
- Cajal, S.R.y. Studies on Vertebrate Neurogenesis; Thomas, C.C., Ed.; Springfield: Virgina, VA, USA, 1960. [Google Scholar]
- Hubel, D.; Wiesel, T.N. Ferrier lecture—Functional architecture of macaque monkey visual cortex. Proc. R. Soc. London Ser. B Biol. Sci. 1977, 198, 1–59. [Google Scholar] [CrossRef]
- Hubel, D.H.; Wiesel, T.N. Receptive fields of single neurones in the cat’s striate cortex. J. Physiol. 1959, 148, 574–591. [Google Scholar] [CrossRef]
- Hubel, D.; Wiesel, T. David Hubel and Torsten Wiesel. Neuron 2012, 75, 182–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubel, D.H.; Wiesel, T.N. Brain Mechanisms of Vision. Sci. Am. 1979, 241, 150–162. [Google Scholar] [CrossRef]
- Sakmann, B.; Neher, E. Patch Clamp Techniques for Studying Ionic Channels in Excitable Membranes. Annu. Rev. Physiol. 1984, 46, 455–472. [Google Scholar] [CrossRef]
- Squire, L.R. Memory for relations in the short term and the long term after medial temporal lobe damage. Hippocampus 2017, 27, 608–612. [Google Scholar] [CrossRef] [Green Version]
- Squire, L.R. On the course of forgetting in very long-term memory. J. Exp. Psychol. Learn. Mem. Cogn. 1989, 15, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.; Slater, P.; Chace, P. Retrograde amnesia: Temporal gradient in very long term memory following electroconvulsive therapy. Science 1975, 187, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G. A Brief History of Epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a018200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, W.J.; Gray, W.R.; Hood, L. The Genetic, Molecular, and Cellular Basis of Antibody Formation: Some Facts and a Unifying Hypothesis. Cold Spring Harb. Symp. Quant. Biol. 1967, 32. [Google Scholar] [CrossRef]
- Gayon, J. From Mendel to epigenetics: History of genetics. Comptes R. Biol. 2016, 339, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Rehen, S.K.; McConnell, M.J.; Kaushal, D.; Kingsbury, M.A.; Yang, A.H.; Chun, J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. USA 2001, 98. [Google Scholar] [CrossRef] [Green Version]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic Copy Number Variation in Human Neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Kingsbury, M.A.; Friedman, B.; McConnell, M.J.; Rehen, S.K.; Yang, A.H.; Kaushal, D.; Chun, J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Natl. Acad. Sci. USA 2005, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehen, S.K. Constitutional Aneuploidy in the Normal Human Brain. J. Neurosci. 2005, 25. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and Confined Chromosomal Mosaicism in the Developing Human Brain. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol. Dis. 2009, 34, 212–220. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Monakhov, V.V.; Soloviev, I.V.; Vostrikov, V.M.; Vorsanova, S.G. The Variation of Aneuploidy Frequency in the Developing and Adult Human Brain Revealed by an Interphase FISH Study. J. Histochem. Cytochem. 2005, 53. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Liehr, T.; Vorsanova, S.G.; Kolotii, A.D.; Yurov, Y.B. Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosom. Res. 2006, 14. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Yurov, Y.B. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. 2009, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushman, D.M.; Chun, J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin. Cell Dev. Biol. 2013, 24, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Yurov, Y.B.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Iourov, I.Y. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 13409–13414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bos, H.; Spierings, D.C.J.; Taudt, A.S.; Bakker, B.; Porubský, D.; Falconer, E.; Novoa, C.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Westra, J.W.; Rivera, R.R.; Bushman, D.M.; Yung, Y.C.; Peterson, S.E.; Barral, S.; Chun, J. Neuronal DNA content variation (DCV) with regional and individual differences in the human brain. J. Comp. Neurol. 2010, 518, 3981–4000. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.G.; Morawski, M.; Brückner, M.K.; Mittag, A.; Tarnok, A.; Arendt, T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 2012, 11, 628–633. [Google Scholar] [CrossRef]
- Bushman, D.M.; Kaeser, G.E.; Siddoway, B.; Westra, J.W.; Rivera, R.R.; Rehen, S.K.; Yung, Y.C.; Chun, J. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. eLife 2015, 4. [Google Scholar] [CrossRef]
- Copani, A.; Hoozemans, J.J.M.; Caraci, F.; Calafiore, M.; Van Haastert, E.S.; Veerhuis, R.; Rozemuller, A.J.M.; Aronica, E.; Sortino, M.A.; Nicoletti, F. DNA Polymerase-beta Is Expressed Early in Neurons of Alzheimer’s Disease Brain and Is Loaded into DNA Replication Forks in Neurons Challenged with beta-Amyloid. J. Neurosci. 2006, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriani, G.A.; Vijg, J.; Montagna, C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 2017, 161. [Google Scholar] [CrossRef] [Green Version]
- Chow, H.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.N.; Potter, H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol. Dis. 1999, 6, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Kingsbury, M.A.; Yung, Y.C.; Peterson, S.E.; Westra, J.W.; Chun, J. Aneuploidy in the normal and diseased brain. Cell. Mol. Life Sci. 2006, 63, 2626–2641. [Google Scholar] [CrossRef]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirifar, P.; Ranjouri, M.R.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr. Allergy Immunol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J. Failed Clearance of Aneuploid Embryonic Neural Progenitor Cells Leads to Excess Aneuploidy in the Atm-Deficient But Not the Trp53-Deficient Adult Cerebral Cortex. J. Neurosci. 2004, 24. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.J. ATM and the Molecular Pathogenesis of Ataxia Telangiectasia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 303–321. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Westra, J.W.; Barral, S.; Chun, J. A Reevaluation of Tetraploidy in the Alzheimer’s Disease Brain. Neurodegener. Dis. 2009, 6. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Brückner, M.K.; Lösche, A. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 2015, 130, 501–510. [Google Scholar] [CrossRef]
- Knouse, K.A.; Wu, J.; Amon, A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 2016, 26, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gole, J.; Gore, A.; Richards, A.; Chiu, Y.J.; Fung, H.L.; Bushman, D.; Chiang, H.I.; Chun, J.; Lo, Y.H.; Zhang, K. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat. Biotechnol. 2013, 31, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chronister, W.D.; Burbulis, I.E.; Wierman, M.B.; Wolpert, M.J.; Haakenson, M.F.; Smith, A.C.B.; Kleinman, J.E.; Hyde, T.M.; Weinberger, D.R.; Bekiranov, S.; et al. Neurons with Complex Karyotypes Are Rare in Aged Human Neocortex. Cell Rep. 2019, 26, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrback, S.; April, C.; Kaper, F.; Rivera, R.R.; Liu, C.S.; Siddoway, B.; Chun, J. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2018, 115, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Lockwood, W.W.; Chari, R.; Chi, B.; Lam, W.L. Recent advances in array comparative genomic hybridization technologies and their applications in human genetics. Eur. J. Hum. Genet. 2006, 14, 139–148. [Google Scholar] [CrossRef]
- Cheung, S.W.; Bi, W. Novel applications of array comparative genomic hybridization in molecular diagnostics. Expert Rev. Mol. Diagn. 2018, 18, 531–542. [Google Scholar] [CrossRef]
- Piotrowski, A.; Bruder, C.E.G.; Andersson, R.; De Ståhl, T.D.; Menzel, U.; Sandgren, J.; Poplawski, A.; Von Tell, D.; Crasto, C.; Bogdan, A.; et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008, 29, 1118–1124. [Google Scholar] [CrossRef]
- Villela, D.; Suemoto, C.K.; Leite, R.; Pasqualucci, C.A.; Grinberg, L.T.; Pearson, P.; Rosenberg, C. Increased DNA Copy Number Variation Mosaicism in Elderly Human Brain. Neural Plast. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Pamphlett, R.; Morahan, J.M.; Luquin, N.; Yu, B. An approach to finding brain-situated mutations in sporadic Parkinson’s disease. Park. Relat. Disord. 2012, 18, 82–85. [Google Scholar] [CrossRef]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-Cell, Genome-wide Sequencing Identifies Clonal Somatic Copy-Number Variation in the Human Brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.E.; Yang, A.H.; Bushman, D.M.; Westra, J.W.; Yung, Y.C.; Barral, S.; Mutoh, T.; Rehen, S.K.; Chun, J. Aneuploid Cells Are Differentially Susceptible to Caspase-Mediated Death during Embryonic Cerebral Cortical Development. J. Neurosci. 2012, 32, 16213–16222. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Abarrategui, J.; Brimblecombe, K.R.; Roberts, R.F.; Velentza-Almpani, E.; Tilley, B.S.; Bengoa-Vergniory, N.; Proukakis, C. Selective vulnerability in α-synucleinopathies. Acta Neuropathol. 2019, 138, 681–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Wenning, G.K.; Colosimo, C.; Geser, F.; Poewe, W. Multiple system atrophy. Lancet Neurol. 2004, 3, 93–103. [Google Scholar] [CrossRef]
- Ahn, T.B.; Kim, S.Y.; Kim, J.Y.; Park, S.S.; Lee, D.S.; Min, H.J.; Kim, Y.K.; Kim, S.E.; Kim, J.M.; Kim, H.J.; et al. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2008, 70, 43–49. [Google Scholar] [CrossRef]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Perandones, C.; Giugni, J.C.; Calvo, D.S.; Raina, G.B.; De Jorge Lopez, L.; Volpini, V.; Zabetian, C.P.; Mata, I.F.; Caputo, M.; Corach, D.; et al. Mosaicism of Alpha-synuclein gene rearrangements: Report of two unrelated cases of early-onset parkinsonism. Park. Relat. Disord. 2014, 20, 558–561. [Google Scholar] [CrossRef] [Green Version]
- Mokretar, K.; Pease, D.; Taanman, J.-W.; Soenmez, A.; Ejaz, A.; Lashley, T.; Ling, H.; Gentleman, S.; Houlden, H.; Holton, J.L.; et al. Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 2018, 141. [Google Scholar] [CrossRef]
- Perez-Rodriguez, D.; Kalyva, M.; Leija-Salazar, M.; Lashley, T.; Tarabichi, M.; Chelban, V.; Gentleman, S.; Schottlaender, L.; Franklin, H.; Vasmatzis, G.; et al. Investigation of somatic CNVs in brains of synucleinopathy cases using targeted SNCA analysis and single cell sequencing. Acta Neuropathol. Commun. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.J.; Wei, W.; Wilson, I.; Coxhead, J.; Ryan, S.; Rollinson, S.; Griffin, H.; Kurzawa-Akanbi, M.; Santibanez-Koref, M.; Talbot, K.; et al. Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res. 2017, 27, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Sala Frigerio, C.; Lau, P.; Troakes, C.; Deramecourt, V.; Gele, P.; Van Loo, P.; Voet, T.; De Strooper, B. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimer’s Dement. 2015, 11, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Pamphlett, R.; Morahan, J.M.; Luquin, N.; Yu, B. Looking for differences in copy number between blood and brain in sporadic amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.; Blauw, H.M.; Veldink, J.H.; van Es, M.A.; Ophoff, R.A.; Bradley, D.G.; van den Berg, L.H.; Hardiman, O. Analysis of genome-wide copy number variation in Irish and Dutch ALS populations. Hum. Mol. Genet. 2008, 17, 3392–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoichet, S.A.; Waibel, S.; Shoichet, S.A.; Waibel, S.; Endruhn, S.; Sperfeld, A.D.; Vorwerk, B.; Müller, I.; Erdogan, F.; Ludolph, A.C.; et al. Identification of candidate genes for sporadic amyotrophic lateral sclerosis by array comparative genomic hybridization. Amyotroph. Lateral Scler. 2009, 10, 162–167. [Google Scholar] [CrossRef]
- Wain, L.V.; Pedroso, I.; Landers, J.E.; Breen, G.; Shaw, C.E.; Leigh, P.N.; Brown, R.H.; Tobin, M.D.; Al-Chalabi, A. The Role of Copy Number Variation in Susceptibility to Amyotrophic Lateral Sclerosis: Genome-Wide Association Study and Comparison with Published Loci. PLoS ONE 2009, 4, e8175. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.; Linton, L.; Birren, B.; Nusbaum, C.; Zody, M.; Baldwin, J. Initial sequencing and analysis of the human genome. Nature 2001, 409. [Google Scholar] [CrossRef] [Green Version]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.A. Human endogenous retroviruses: Friend or foe? APMIS 2016, 124, 4–10. [Google Scholar] [CrossRef]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.J.; Billon, V. L1 retrotransposition in the soma: A field jumping ahead. Mob. DNA 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kines, K.J.; Sokolowski, M.; deHaro, D.L.; Christian, C.M.; Belancio, V.P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Estécio, M.R.H.; Gallegos, J.; Dekmezian, M.; Lu, Y.; Liang, S.; Issa, J.-P.J. SINE Retrotransposons Cause Epigenetic Reprogramming of Adjacent Gene Promoters. Mol. Cancer Res. 2012, 10, 1332–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorek, R. Alu-Containing Exons are Alternatively Spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [Green Version]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which transposable elements are active in the human genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chitiprolu, M.; Gagnon, D.; Meng, L.; Perez-Iratxeta, C.; Lagace, D.; Gibbings, D. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat. Commun. 2014, 5, 5276. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosom. Res. 2018, 26, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Della Valle, F.; Thimma, M.P.; Caiazzo, M.; Pulcrano, S.; Celii, M.; Adroub, S.A.; Liu, P.; Alanis-Lobato, G.; Broccoli, V.; Orlando, V. Transdifferentiation of Mouse Embryonic Fibroblasts into Dopaminergic Neurons Reactivates LINE-1 Repetitive Elements. Stem Cell Rep. 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Bedrosian, T.A.; Quayle, C.; Novaresi, N.; Gage, F.H. Early life experience drives structural variation of neural genomes in mice. Science 2018, 359. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; del-Pozo-Martín, Y.; Carrión, Á.M. L1 retrotransposition alters the hippocampal genomic landscape enabling memory formation. Brain Behav. Immun. 2017, 64. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460. [Google Scholar] [CrossRef] [Green Version]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-Neuron Sequencing Analysis of L1 Retrotransposition and Somatic Mutation in the Human Brain. Cell 2012, 151. [Google Scholar] [CrossRef] [Green Version]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sánchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 Mosaicism in Hippocampal Neurons. Cell 2015, 161. [Google Scholar] [CrossRef] [Green Version]
- Evrony, G.D.; Lee, E.; Park, P.J.; Walsh, C.A. Resolving rates of mutation in the brain using single-neuron genomics. eLife 2016, 5. [Google Scholar] [CrossRef]
- Padmanabhan Nair, V.; Liu, H.; Ciceri, G.; Jungverdorben, J.; Frishman, G.; Tchieu, J.; Cederquist, G.Y.; Rothenaigner, I.; Schorpp, K.; Klepper, L.; et al. Activation of HERV-K(HML-2) disrupts cortical patterning and neuronal differentiation by increasing NTRK3. Cell Stem Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Erwin, J.A.; Paquola, A.C.M.; Singer, T.; Gallina, I.; Novotny, M.; Quayle, C.; Bedrosian, T.A.; Alves, F.I.A.; Butcher, C.R.; Herdy, J.R.; et al. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat. Neurosci. 2016, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, S.; Seleme, M.d.C.; Soifer, H.S.; Perez, J.L.G.; Moran, J.V.; Kazazian, H.H.; Kasahara, N. L1 retrotransposition in nondividing and primary human somatic cells. Proc. Natl. Acad. Sci. USA 2006, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Mates, L.; Ivics, Z.; Izsvák, Z.; Martin, S.L.; An, W. Cell division promotes efficient retrotransposition in a stable L1 reporter cell line. Mob. DNA 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanikar, J.N. A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 2004, 32. [Google Scholar] [CrossRef] [Green Version]
- Yang, N. An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 2003, 31. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.N.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Garcia, E.; Moreno-Cugnon, L.; Garcia, I.; Borras, C.; Revuelta, M.; Izeta, A.; Lopez-Lluch, G.; de Pancorbo, M.M.; Vergara, I.; Vina, J.; et al. SOX2 expression diminishes with ageing in several tissues in mice and humans. Mech. Ageing Dev. 2019, 177, 30–36. [Google Scholar] [CrossRef]
- Sarlak, G.; Vincent, B. The Roles of the Stem Cell-Controlling Sox2 Transcription Factor: From Neuroectoderm Development to Alzheimer’s Disease? Mol. Neurobiol. 2016, 53, 1679–1698. [Google Scholar] [CrossRef]
- Macia, A.; Widmann, T.J.; Heras, S.R.; Ayllon, V.; Sanchez, L.; Benkaddour-Boumzaouad, M.; Muñoz-Lopez, M.; Rubio, A.; Amador-Cubero, S.; Blanco-Jimenez, E.; et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017, 27, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Bundo, M.; Kato, T.; Iwamoto, K. Estimation of LINE-1 Copy Number in the Brain Tissue and Isolated Neuronal Nuclei. In Genomic Mosaicism in Neurons and Other Cell Types; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.T.; Faulkner, G.J.; Dubnau, J.; Ponomarev, I.; Gage, F.H. The Role of Transposable Elements in Health and Diseases of the Central Nervous System. J. Neurosci. 2013, 33, 17577–17586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21. [Google Scholar] [CrossRef] [Green Version]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Carson, C.T.; Macia, A.; Moran, J.V.; Gage, F.H. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob-Hirsch, J.; Eyal, E.; Knisbacher, B.A.; Roth, J.; Cesarkas, K.; Dor, C.; Farage-Barhom, S.; Kunik, V.; Simon, A.J.; Gal, M.; et al. Whole-genome sequencing reveals principles of brain retrotransposition in neurodevelopmental disorders. Cell Res. 2018, 28. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23. [Google Scholar] [CrossRef]
- Zhao, B.; Wu, Q.; Ye, A.Y.; Guo, J.; Zheng, X.; Yang, X.; Yan, L.; Liu, Q.-R.; Hyde, T.M.; Wei, L.; et al. Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25. [Google Scholar] [CrossRef] [Green Version]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Steele, A.J.; Al-Chalabi, A.; Ferrante, K.; Cudkowicz, M.E.; Brown, R.H.; Garson, J.A. Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 2005, 64. [Google Scholar] [CrossRef]
- McCormick, A.L.; Brown, R.H.; Cudkowicz, M.E.; Al-Chalabi, A.; Garson, J.A. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 2008, 70. [Google Scholar] [CrossRef] [PubMed]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Harris, B.T.; Ostrow, L.W.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.Y.; Russ, J.; Cali, C.P.; Phan, J.M.; Amlie-Wolf, A.; Lee, E.B. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019, 27. [Google Scholar] [CrossRef] [Green Version]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Harz, C.; Sanchez, L.; Pereira, G.C.; Maldener, E.; Heras, S.R.; Ostrow, L.W.; Ravits, J.; Batra, R.; Meese, E.; et al. Transcriptional profiling of HERV-K(HML-2) in amyotrophic lateral sclerosis and potential implications for expression of HML-2 proteins. Mol. Neurodegener. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23. [Google Scholar] [CrossRef]
- Protasova, M.S.; Gusev, F.E.; Grigorenko, A.P.; Kuznetsova, I.L.; Rogaev, E.I.; Andreeva, T.V. Quantitative Analysis of L1 Retrotransposons in Alzheimer’s Disease and Aging. Biochem. Mosc. 2017, 82, 962–971. [Google Scholar] [CrossRef]
- Khristich, A.N.; Mirkin, S.M. On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar] [CrossRef] [Green Version]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef]
- Kovtun, I.V.; McMurray, C.T. Trinucleotide expansion in haploid germ cells by gap repair. Nat. Genet. 2001, 27, 407–411. [Google Scholar] [CrossRef]
- Kennedy, L.; Evans, E.; Chen, C.M.; Craven, L.; Detloff, P.J.; Ennis, M.; Shelbourne, P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003, 12, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Swami, M.; Hendricks, A.E.; Gillis, T.; Massood, T.; Mysore, J.; Myers, R.H.; Wheeler, V.C. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 2009, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telenius, H.; Kremer, B.; Goldberg, Y.P.; Theilmann, J.; Andrew, S.E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E.J.; Clarke, L.A.; et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Shelbourne, P.F.; Keller-McGandy, C.; Bi, W.L.; Yoon, S.R.; Dubeau, L.; Veitch, N.J.; Vonsattel, J.P.; Wexler, N.S.; Group, U.S.-V.C.R.; Arnheim, N.; et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet. 2007, 16, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Hubert, L., Jr.; Wilson, J.H. Transcription destabilizes triplet repeats. Mol. Carcinog. 2009, 48, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Petruska, J.; Hartenstine, M.J.; Goodman, M.F. Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. J. Biol. Chem. 1998, 273, 5204–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M.; et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Chao, M.J.; Harold, D.; Abu Elneel, K.; Gillis, T.; Holmans, P.; Jones, L.; Orth, M.; Myers, R.H.; Kwak, S.; et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet. 2017, 26, 3859–3867. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.J.H.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; investigators, T.-H.; investigators, R.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Tome, S.; Manley, K.; Simard, J.P.; Clark, G.W.; Slean, M.M.; Swami, M.; Shelbourne, P.F.; Tillier, E.R.; Monckton, D.G.; Messer, A.; et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013, 9, e1003280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Tejwani, L.; Lim, J. Pathogenic mechanisms underlying spinocerebellar ataxia type 1. Cell. Mol. Life Sci. 2020, 77, 4015–4029. [Google Scholar] [CrossRef] [PubMed]
- Kraus-Perrotta, C.; Lagalwar, S. Expansion, mosaicism and interruption: Mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1. Cerebellum Ataxias 2016, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Mouro Pinto, R.; Arning, L.; Giordano, J.V.; Razghandi, P.; Andrew, M.A.; Gillis, T.; Correia, K.; Mysore, J.S.; Grote Urtubey, D.-M.; Parwez, C.R.; et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2012, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendron, T.F.; Petrucelli, L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a024224. [Google Scholar] [CrossRef] [Green Version]
- Babić Leko, M.; Župunski, V.; Kirincich, J.; Smilović, D.; Hortobágyi, T.; Hof, P.R.; Šimić, G. Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion. Behav. Neurol. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, V.L.; Cooper-Knock, J.; Connor-Robson, N.; Higginbottom, A.; Kirby, J.; Razinskaya, O.D.; Ninkina, N.; Shaw, P.J. Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol. Neurodegener. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dols-Icardo, O.; García-Redondo, A.; Rojas-García, R.; Sánchez-Valle, R.; Noguera, A.; Gómez-Tortosa, E.; Pastor, P.; Hernández, I.; Esteban-Pérez, J.; Suárez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Nordin, A.; Akimoto, C.; Wuolikainen, A.; Alstermark, H.; Jonsson, P.; Birve, A.; Marklund, S.L.; Graffmo, K.S.; Forsberg, K.; Brännström, T.; et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 2015, 24, 3133–3142. [Google Scholar] [CrossRef]
- Ross, J.P.; Leblond, C.S.; Catoire, H.; Volkening, K.; Strong, M.; Zinman, L.; Robertson, J.; Dion, P.A.; Rouleau, G.A. Somatic expansion of the C9orf72 hexanucleotide repeat does not occur in ALS spinal cord tissues. Neurol. Genet. 2019, 5, e317. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Hozumi, N.; Tonegawa, S. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc. Natl. Acad. Sci. USA 1976, 73, 3628–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-H.; Liu, C.S.; Zhu, Y.; Kaeser, G.E.; Rivera, R.; Romanow, W.J.; Kihara, Y.; Chun, J. Reply to: APP gene copy number changes reflect exogenous contamination. Nature 2020, 584, E29–E33. [Google Scholar] [CrossRef] [PubMed]
- Pacific Biosciences Data Release: Alzheimer Brain Isoform Sequencing (Iso-Seq) Dataset; Pacific Biosciences: Menlo Park, CA, USA, 2016.
- Park, J.S.; Lee, J.; Jung, E.S.; Kim, M.H.; Kim, I.B.; Son, H.; Kim, S.; Kim, S.; Park, Y.M.; Mook-Jung, I.; et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zhao, B.; Huang, A.Y.; Miller, M.B.; Lodato, M.A.; Walsh, C.A.; Lee, E.A. APPgene copy number changes reflect exogenous contamination. Nature 2020, 584, E20–E28. [Google Scholar] [CrossRef]
- Kaeser, G.; Chun, J. Brain cell somatic gene recombination and its phylogenetic foundations. J. Biol. Chem. 2020, 295, 12786–12795. [Google Scholar] [CrossRef] [PubMed]
- Hooli, B.V.; Kovacs-Vajna, Z.M.; Mullin, K.; Blumenthal, M.A.; Mattheisen, M.; Zhang, C.; Lange, C.; Mohapatra, G.; Bertram, L.; Tanzi, R.E. Rare autosomal copy number variations in early-onset familial Alzheimer’s disease. Mol. Psychiatry 2014, 19, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Kaeser, G.E.; Chun, J. Mosaic Somatic Gene Recombination as a Potentially Unifying Hypothesis for Alzheimer’s Disease. Front. Genet. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.A. Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 1725–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, T.; Tomasini, L.; Mariani, J.; Zhou, B.; Roychowdhury, T.; Franjic, D.; Pletikos, M.; Pattni, R.; Chen, B.J.; Venturini, E.; et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 2018, 359, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Keogh, M.J.; Aryaman, J.; Golder, Z.; Kullar, P.J.; Wilson, I.; Talbot, K.; Turner, M.R.; McKenzie, C.A.; Troakes, C.; et al. Frequency and signature of somatic variants in 1461 human brain exomes. Genet. Med. 2019, 21, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nieto, P.E.; Morrison, A.J.; Fraser, H.B. The somatic mutation landscape of the human body. Genome Biol. 2019, 20, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Hoang, M.L.; Kinde, I.; Tomasetti, C.; McMahon, K.W.; Rosenquist, T.A.; Grollman, A.P.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2016, 113, 9846–9851. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364. [Google Scholar] [CrossRef]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Uphill, J.B.; Adamson, G.; Geddes, J.F.; Revesz, T.; Davis, M.B.; Wood, N.W.; Collinge, J.; et al. Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Hisahara, S.; Nishiyama, A.; Tsuda, E.; Suzuki, S.; Matsumura, A.; Ishikawa, A.; Sakurai, A.; Motoike, I.N.; Aoki, M.; Aoki, Y.; et al. Possible Somatic Mosaicism of Novel FUS Variant in Familial Amyotrophic Lateral Sclerosis. Neurol. Genet. 2021, 7, e552. [Google Scholar] [CrossRef]
- Parcerisas, A.; Rubio, S.E.; Muhaisen, A.; Gómez-Ramos, A.; Pujadas, L.; Puiggros, M.; Rossi, D.; Ureña, J.; Burgaya, F.; Pascual, M.; et al. Somatic Signature of Brain-Specific Single Nucleotide Variations in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 1357–1382. [Google Scholar] [CrossRef] [Green Version]
- Leija-Salazar, M.; Pittman, A.; Mokretar, K.; Morris, H.; Schapira, A.H.; Proukakis, C. Investigation of Somatic Mutations in Human Brains Targeting Genes Associated With Parkinson’s Disease. Front. Neurol. 2020, 11, 8–12. [Google Scholar] [CrossRef]
- Proukakis, C.; Houlden, H.; Schapira, A.H. Somatic Alpha-synuclein mutations in Parkinson’s disease: Hypothesis and preliminary data. Mov. Disord. 2013, 28, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Proukakis, C.; Shoaee, M.; Morris, J.; Brier, T.; Kara, E.; Sheerin, U.M.; Charlesworth, G.; Tolosa, E.; Houlden, H.; Wood, N.W.; et al. Analysis of Parkinson’s disease brain-derived DNA for Alpha-synuclein coding somatic mutations. Mov. Disord. 2014, 29, 1060–1064. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.J.; Wei, W.; Aryaman, J.; Walker, L.; van den Ameele, J.; Coxhead, J.; Wilson, I.; Bashton, M.; Beck, J.; West, J.; et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Acuña-Hidalgo, R.; Keogh, M.J.; Quenez, O.; Steehouwer, M.; Lelieveld, S.; Rousseau, S.; Richard, A.C.; Oud, M.S.; Marguet, F.; et al. Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.T.; Lundin, P.; Wallén Arzt, E.; Lindström, A.K.; Graff, C.; Eriksson, M. Somatic mutation that affects transcription factor binding upstream of CD55 in the temporal cortex of a late-onset Alzheimer disease patient. Hum. Mol. Genet. 2019, 28, 2675–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Yang, L.; Li, R. What does complement do in Alzheimer’s disease? Old molecules with new insights. Transl. Neurodegener. 2013, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashko-Pachima, Y.; Hadar, A.; Grigg, I.; Korenková, V.; Kapitansky, O.; Karmon, G.; Gershovits, M.; Sayas, C.L.; Kooy, R.F.; Attems, J.; et al. Discovery of autism/intellectual disability somatic mutations in Alzheimer’s brains: Mutated ADNP cytoskeletal impairments and repair as a case study. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [Green Version]
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Y.; Gold, H.D.; Luquette, L.J.; Park, P.J. Detecting Somatic Mutations in Normal Cells. Trends Genet. 2018, 34. [Google Scholar] [CrossRef]
- Wang, K.; Lai, S.; Yang, X.; Zhu, T.; Lu, X.; Wu, C.-I.; Ruan, J. Ultrasensitive and high-efficiency screen of de novo low-frequency mutations by o2n-seq. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, E.H.; Lee, S.H. Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing. Int. J. Mol. Sci. 2019, 20, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, D.; Lim, J.S.; Maeng, J.H.; Son, H.; Kang, H.-C.; Nam, H.; Lee, J.H.; Kim, S. The use of technical replication for detection of low-level somatic mutations in next-generation sequencing. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Reiner, B.C.; Doyle, G.A.; Weller, A.E.; Levinson, R.N.; Namoglu, E.; Pigeon, A.; Perea, E.D.; Weickert, C.S.; Turecki, G.; Mash, D.C.; et al. Restriction Enzyme Based Enriched L1Hs Sequencing (REBELseq): A Scalable Technique for Detection of Ta Subfamily L1Hs in the Human Genome. G3 Genes Genomes Genet. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Doan, R.N.; Miller, M.B.; Kim, S.N.; Rodin, R.E.; Ganz, J.; Bizzotto, S.; Morillo, K.S.; Huang, A.Y.; Digumarthy, R.; Zemmel, Z.; et al. MIPP-Seq: Ultra-sensitive rapid detection and validation of low-frequency mosaic mutations. BMC Med. Genom. 2021, 14. [Google Scholar] [CrossRef]
- Cooke, S.L.; Shlien, A.; Marshall, J.; Pipinikas, C.P.; Martincorena, I.; Tubio, J.M.C.; Li, Y.; Menzies, A.; Mudie, L.; Ramakrishna, M.; et al. Processed pseudogenes acquired somatically during cancer development. Nat. Commun. 2014, 5, 3644. [Google Scholar] [CrossRef]
- Henssen, A.G.; Koche, R.; Zhuang, J.; Jiang, E.; Reed, C.; Eisenberg, A.; Still, E.; MacArthur, I.C.; Rodríguez-Fos, E.; Gonzalez, S.; et al. PGBD5 promotes site-specific oncogenic mutations in human tumors. Nat. Genet. 2017, 49, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Siudeja, K.; Beek, M.; Riddiford, N.; Boumard, B.; Wurmser, A.; Stefanutti, M.; Lameiras, S.; Bardin, A.J. Unraveling the features of somatic transposition in the Drosophila intestine. EMBO J. 2021, 40. [Google Scholar] [CrossRef]
- Davie, K.; Janssens, J.; Koldere, D.; De Waegeneer, M.; Pech, U.; Kreft, Ł.; Aibar, S.; Makhzami, S.; Christiaens, V.; Bravo González-Blas, C.; et al. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 2018, 174, 982–998.e20. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.D. The Genome Sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [Green Version]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Herculano-Houzel, S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc. Natl. Acad. Sci. USA 2012, 109, 10661–10668. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Turner, K.M.; Nguyen, N.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Møller, H.D.; Mohiyuddin, M.; Prada-Luengo, I.; Sailani, M.R.; Halling, J.F.; Plomgaard, P.; Maretty, L.; Hansen, A.J.; Snyder, M.P.; Pilegaard, H.; et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 2018, 9, 1069. [Google Scholar] [CrossRef] [Green Version]
- Morton, A.R.; Dogan-Artun, N.; Faber, Z.J.; MacLeod, G.; Bartels, C.F.; Piazza, M.S.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019, 179, 1330–1341.e13. [Google Scholar] [CrossRef] [PubMed]
- Koche, R.P.; Rodriguez-Fos, E.; Helmsauer, K.; Burkert, M.; MacArthur, I.C.; Maag, J.; Chamorro, R.; Munoz-Perez, N.; Puiggròs, M.; Dorado Garcia, H.; et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 2020, 52, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Kumar, P.; Layer, R.; Willcox, S.; Gagan, J.R.; Griffith, J.D.; Dutta, A. Extrachromosomal MicroDNAs and Chromosomal Microdeletions in Normal Tissues. Science 2012, 336, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.-H.; Zheng, X.; Fu, M.; Huang, C.; Yang, X. The decreased exclusion of nuclear eccDNA: From molecular and subcellular levels to human aging and age-related diseases. Ageing Res. Rev. 2021, 67, 101306. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.; Schmeer, C.; Wengerodt, D.; Witte, O.W.; Kretz, A. Extrachromosomal Circular DNA: Current Knowledge and Implications for CNS Aging and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, V.; Luebeck, J.; Nguyen, N.-P.D.; Bakhtiari, M.; Turner, K.M.; Schwab, R.; Carter, H.; Mischel, P.S.; Bafna, V. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 2019, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.-C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143. [Google Scholar] [CrossRef]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Hornung, S.; Dutta, S.; Bitan, G. CNS-Derived Blood Exosomes as a Promising Source of Biomarkers: Opportunities and Challenges. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costantino, I.; Nicodemus, J.; Chun, J. Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain. Genes 2021, 12, 1071. https://doi.org/10.3390/genes12071071
Costantino I, Nicodemus J, Chun J. Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain. Genes. 2021; 12(7):1071. https://doi.org/10.3390/genes12071071
Chicago/Turabian StyleCostantino, Isabel, Juliet Nicodemus, and Jerold Chun. 2021. "Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain" Genes 12, no. 7: 1071. https://doi.org/10.3390/genes12071071
APA StyleCostantino, I., Nicodemus, J., & Chun, J. (2021). Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain. Genes, 12(7), 1071. https://doi.org/10.3390/genes12071071