
Nuclear Organization during Hepatogenesis in Zebrafish Requires Uhrf1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry and Genotyping
2.2. RNA and DNA Extraction
2.3. cDNA Production and qPCR
2.4. RNA-Seq
2.5. ATAC-Seq
2.6. Reduced-Representation Bisulfite Sequencing
2.7. Bioinformatic Analysis
2.8. Immunofluorescence
2.9. Confocal Imaging, Image Processing, and Analysis
2.10. Statistical Analysis
3. Results
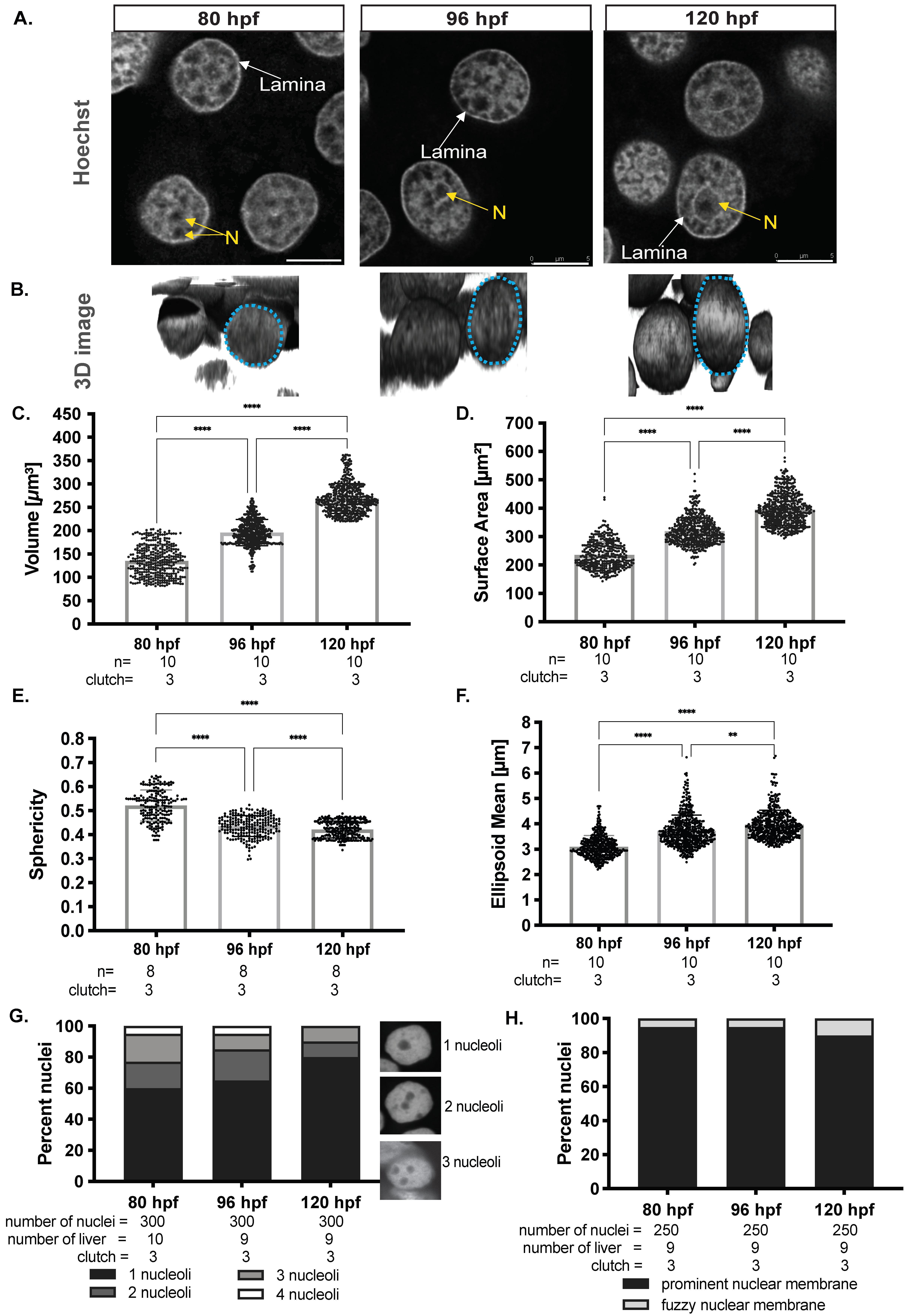
3.1. Hepatocyte Nuclear Morphology Evolves during Hepatic Outgrowth
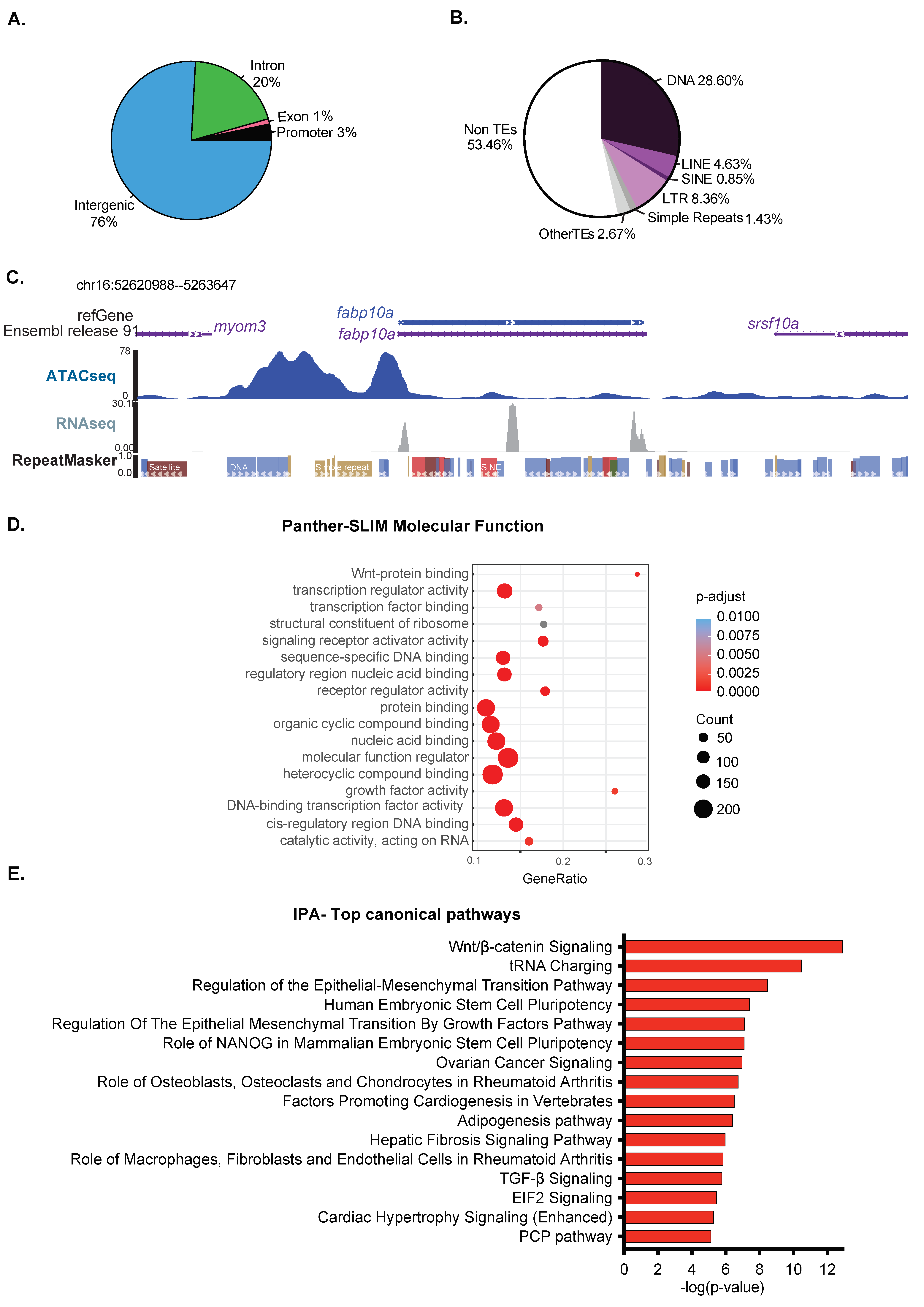
3.2. Open Chromatin in the Larval Liver Is Enriched for Developmental Genes
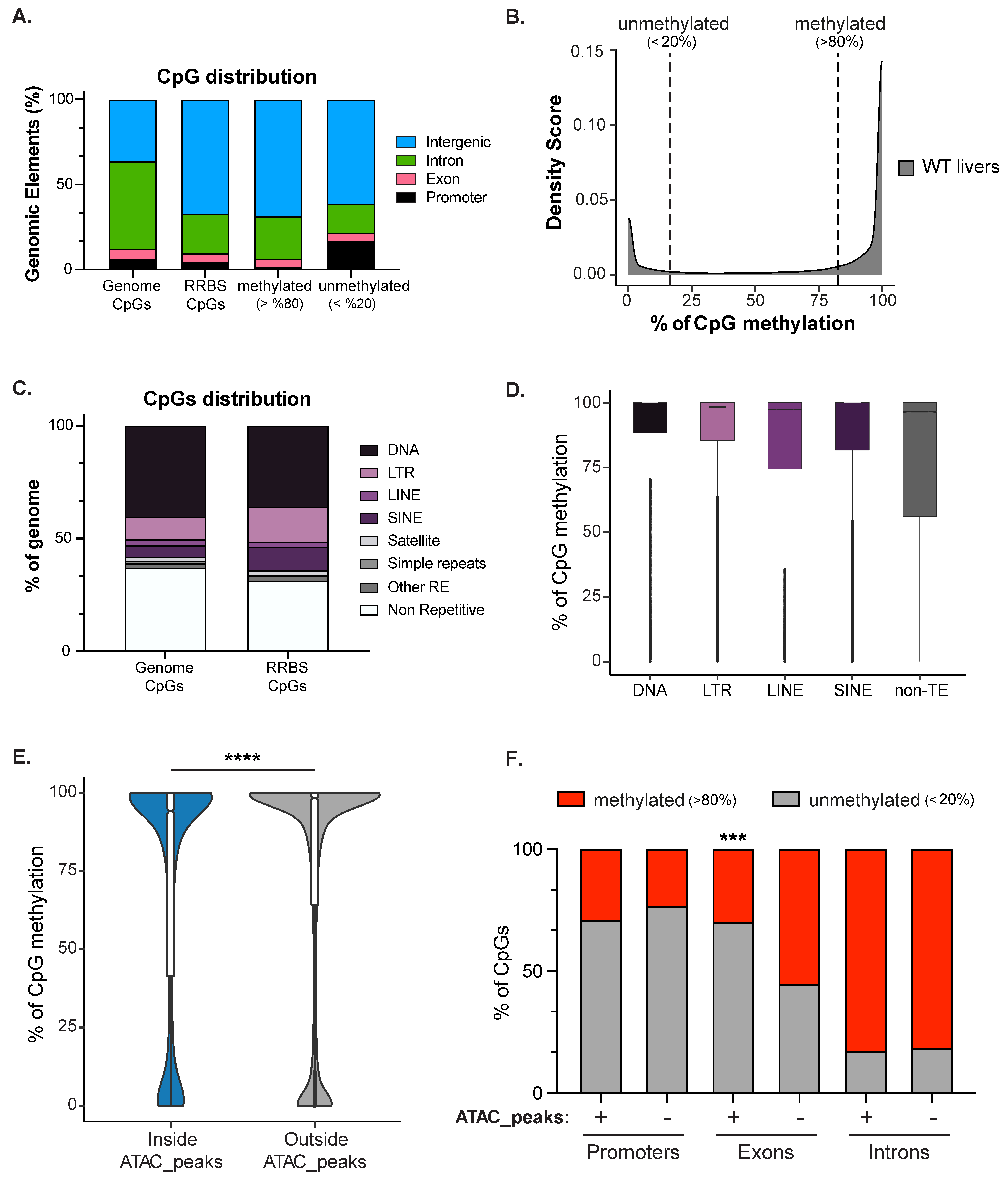
3.3. Hepatic DNA Methylome Is Enriched in the Intergenome and on Transposons
3.4. Transcriptome of the Late Developing Liver Reflects Hepatic Maturation
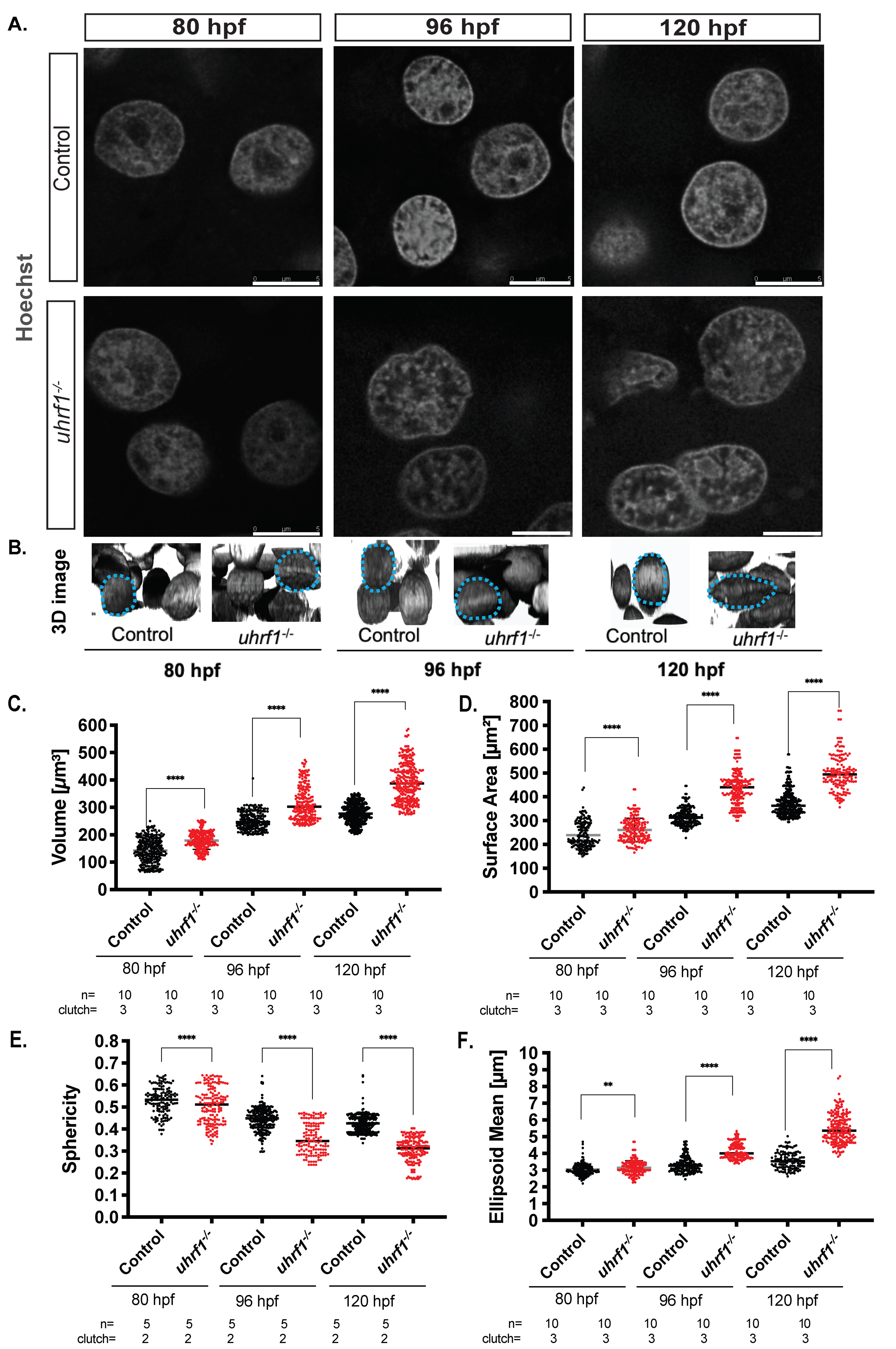
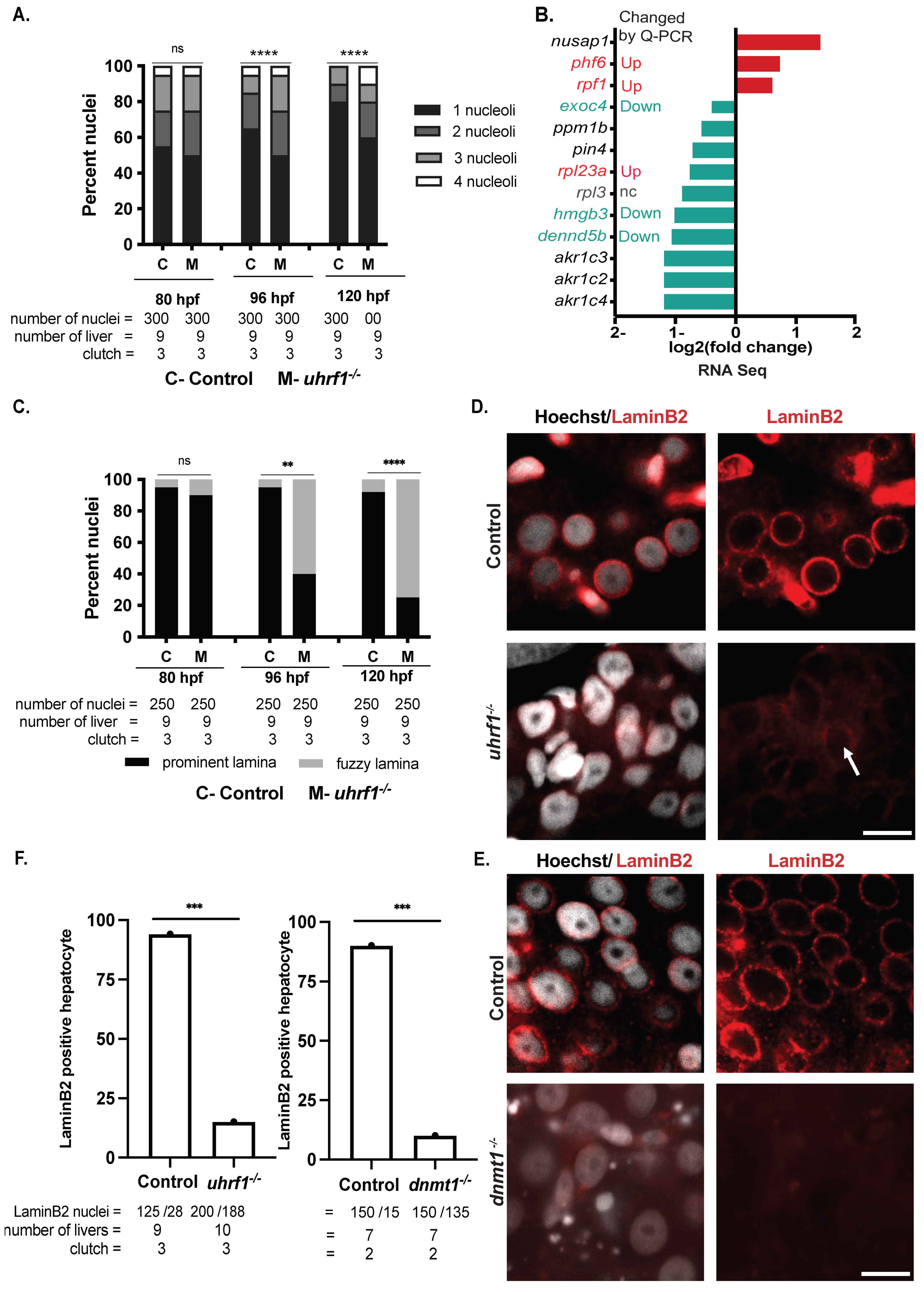
3.5. Uhrf1 Loss Leads to Large Dysmorphic Hepatocyte Nuclei
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Foreman, R.; Wollman, R. Identifying chromatin features that regulate gene expression distribution. Sci. Rep. 2020, 10, 20566. [Google Scholar] [CrossRef] [PubMed]
- Moindrot, B.; Bouvet, P.; Mongelard, F. Chromatin structure and organization: The relation with gene expression during development and disease. Subcell Biochem. 2013, 61, 373–396. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 772. [Google Scholar] [CrossRef]
- Madakashira, B.P.; Sadler, K.C. DNA Methylation, Nuclear Organization, and Cancer. Front. Genet. 2017, 8, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Blumenthal, R.M. Coordinated chromatin control: Structural and functional linkage of DNA and histone methylation. Biochemistry 2010, 49, 2999–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Galiova, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ali, M.; Zhou, Q. Establishment and evolution of heterochromatin. Ann. N. Y. Acad. Sci. 2020, 1476, 59–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, B.M.; Johnson, E.E. Nuclear morphologies: Their diversity and functional relevance. Chromosoma 2017, 126, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fussner, E.; Djuric, U.; Strauss, M.; Hotta, A.; Perez-Iratxeta, C.; Lanner, F.; Dilworth, F.J.; Ellis, J.; Bazett-Jones, D.P. Constitutive heterochromatin reorganization during somatic cell reprogramming. EMBO J. 2011, 30, 1778–1789. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Veerapandian, V.; Yang, X.; Song, K.; Xu, X.; Cui, M.; Yuan, W.; Huang, Y.; Xia, X.; Yao, Z.; et al. The chromatin accessibility landscape reveals distinct transcriptional regulation in the induction of human primordial germ cell-like cells from pluripotent stem cells. Stem Cell Rep. 2021, 16, 1245–1261. [Google Scholar] [CrossRef]
- Guertin, M.J.; Lis, J.T. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr. Opin. Genet. Dev. 2013, 23, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laue, K.; Rajshekar, S.; Courtney, A.J.; Lewis, Z.A.; Goll, M.G. The maternal to zygotic transition regulates genome-wide heterochromatin establishment in the zebrafish embryo. Nat. Commun. 2019, 10, 1551. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.L.; Duronio, R.J. Phasing in heterochromatin during development. Genes Dev. 2019, 33, 379–381. [Google Scholar] [CrossRef]
- Mancini, M.; Magnani, E.; Macchi, F.; Bonapace, I.M. The multi-functionality of UHRF1: Epigenome maintenance and preservation of genome integrity. Nucleic Acids Res. 2021, 49, 6053–6068. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Q.; Li, P.; Zhao, Q.; Zhang, J.; Li, J.; Koseki, H.; Wong, J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013, 4, 1563. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.S.; Cornett, E.M.; Goldfarb, D.; DaRosa, P.A.; Li, Z.M.; Yan, F.; Dickson, B.M.; Guo, A.H.; Cantu, D.V.; Kaustov, L.; et al. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, A.; Yamaguchi, L.; Sharif, J.; Johmura, Y.; Kawamura, T.; Nakanishi, K.; Shimamura, S.; Arita, K.; Kodama, T.; Ishikawa, F.; et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013, 502, 249–253. [Google Scholar] [CrossRef]
- Citterio, E.; Papait, R.; Nicassio, F.; Vecchi, M.; Gomiero, P.; Mantovani, R.; Di Fiore, P.P.; Bonapace, I.M. Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol. Cell Biol. 2004, 24, 2526–2535. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forne, I.; Pichler, G.; Horl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Tsai, H.C.; Yen, R.C.; Zhang, Y.W.; Kong, X.; Wang, W.; Xia, L.; Baylin, S.B. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017, 27, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Esteve, P.O.; Jacobsen, S.E.; Pradhan, S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009, 37, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Chernyavskaya, Y.; Mudbhary, R.; Zhang, C.; Tokarz, D.; Jacob, V.; Gopinath, S.; Sun, X.; Wang, S.; Magnani, E.; Madakashira, B.P.; et al. Loss of DNA methylation in zebrafish embryos activates retrotransposons to trigger antiviral signaling. Development 2017, 144, 2925–2939. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Loughlin, E.A.; Gaur, N.A.; SenBanerjee, S.; Jacob, V.; Monson, C.; Kent, B.; Oranu, A.; Ding, Y.; Ukomadu, C.; et al. UHRF1 phosphorylation by cyclin A2/cyclin-dependent kinase 2 is required for zebrafish embryogenesis. Mol. Biol. Cell 2012, 23, 59–70. [Google Scholar] [CrossRef]
- Jacob, V.; Chernyavskaya, Y.; Chen, X.; Tan, P.S.; Kent, B.; Hoshida, Y.; Sadler, K.C. DNA hypomethylation induces a DNA replication-associated cell cycle arrest to block hepatic outgrowth in uhrf1 mutant zebrafish embryos. Development 2015, 142, 510–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, B.; Magnani, E.; Walsh, M.J.; Sadler, K.C. UHRF1 regulation of Dnmt1 is required for pre-gastrula zebrafish development. Dev. Biol. 2016, 412, 99–113. [Google Scholar] [CrossRef]
- Magnani, E.; Macchi, F.; Madakashira, B.P.; Zhang, C.; Alaydaroos, F.; Sadler, K.C. uhrf1 and dnmt1 Loss Induces an Immune Response in Zebrafish Livers Due to Viral Mimicry by Transposable Elements. Front. Immunol. 2021, 12, 627926. [Google Scholar] [CrossRef] [PubMed]
- Tien, A.L.; Senbanerjee, S.; Kulkarni, A.; Mudbhary, R.; Goudreau, B.; Ganesan, S.; Sadler, K.C.; Ukomadu, C. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem. J. 2011, 435, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Wang, J.; Gong, W.; Zhang, M.; Tang, Z.; Zhang, J.; Quan, Z. UHRF1 depletion suppresses growth of gallbladder cancer cells through induction of apoptosis and cell cycle arrest. Oncol. Rep. 2014, 31, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Chen, Y.; Yang, X.; Wang, P.; Liu, T.; Deng, M.; Qin, B.; Correia, C.; Lee, S.; et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 2016, 7, 10201. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, E.A.; Lemaigre, F.P. Development of the liver: Insights into organ and tissue morphogenesis. J. Hepatol. 2018, 68, 1049–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Miller, S.R.; Ober, E.A.; Sadler, K.C. Making It New Again: Insight Into Liver Development, Regeneration, and Disease From Zebrafish Research. Curr. Top. Dev. Biol. 2017, 124, 161–195. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; Stainier, D.Y. Endoderm specification and liver development. Methods Cell Biol. 2016, 134, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Monga, S.P. Cellular and molecular basis of liver development. Compr. Physiol. 2013, 3, 799–815. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Al-Diffhala, S.; Centeno, B.A. Primary Liver Cancers-Part 1: Histopathology, Differential Diagnoses, and Risk Stratification. Cancer Control. 2018, 25, 1073274817744625. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Functional anatomy of normal bile ducts. Anat. Rec. 2008, 291, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Chomphuwiset, P.; Magee, D.; Boyle, R.; Treanor, D. Context-Based Classification of Cell nuclei and Tissue Regions in Liver Histopathology. MIUA 2011, 2011, 239–244. [Google Scholar]
- Rao, R.K.; Samak, G. Bile duct epithelial tight junctions and barrier function. Tissue Barriers 2013, 1, e25718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, A. Nucleus, Nuclear Structure, and Nuclear Functional Changes in Liver Cancer. In Tumors and Tumor-Like Lesions of the Hepatobiliary Tract: General and Surgical Pathology; Springer International Publishing: Cham, Switzerland, 2017; pp. 3043–3069. [Google Scholar] [CrossRef]
- Stepinski, D. The nucleolus, an ally, and an enemy of cancer cells. Histochem. Cell Biol. 2018, 150, 607–629. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Sadler, K.C. New school in liver development: Lessons from zebrafish. Hepatology 2009, 50, 1656–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goessling, W.; Sadler, K.C. Zebrafish: An Important Tool for Liver Disease Research. Gastroenterology 2015, 149, 1361–1377. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.H.; Zhang, C.; Yin, C. Using zebrafish to model liver diseases-Where do we stand? Curr. Pathobiol. Rep. 2017, 5, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Luan, Y.; Liu, T.; Lee, H.J.; Fang, L.; Wang, Y.; Wang, X.; Zhang, B.; Jin, Q.; Ang, K.C.; et al. A map of cis-regulatory elements and 3D genome structures in zebrafish. Nature 2020, 588, 337–343. [Google Scholar] [CrossRef]
- Sadler, K.C.; Krahn, K.N.; Gaur, N.A.; Ukomadu, C. Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, uhrf1. Proc. Natl. Acad. Sci. USA 2007, 104, 1570–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amsterdam, A.; Nissen, R.M.; Sun, Z.; Swindell, E.C.; Farrington, S.; Hopkins, N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 2004, 101, 12792–12797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tittle, R.K.; Sze, R.; Ng, A.; Nuckels, R.J.; Swartz, M.E.; Anderson, R.M.; Bosch, J.; Stainier, D.Y.; Eberhart, J.K.; Gross, J.M. Uhrf1 and Dnmt1 are required for development and maintenance of the zebrafish lens. Dev. Biol. 2011, 350, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Cheung, I.D.; Bagnat, M.; Ma, T.P.; Datta, A.; Evason, K.; Moore, J.C.; Lawson, N.D.; Mostov, K.E.; Moens, C.B.; Stainier, D.Y. Regulation of intrahepatic biliary duct morphogenesis by Claudin 15-like b. Dev. Biol. 2012, 361, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; Lopez Pazmino Mdel, P.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y. Identification of Chemical Inhibitors of β-Catenin-Driven Liver Tumorigenesis in Zebrafish. PLoS Genet. 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Bambino, K.; Zhang, C.; Austin, C.; Amarasiriwardena, C.; Arora, M.; Chu, J.; Sadler, K.C. Inorganic arsenic causes fatty liver and interacts with ethanol to cause alcoholic liver disease in zebrafish. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Liu, H.; Yu, J.; Kelliher, M.A.; Castilla, L.H.; Lawson, N.D.; Zhu, L.J. ATACseqQC: A Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genome 2018, 19, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett-Bakelman, F.E.; Sheridan, C.K.; Kacmarczyk, T.J.; Ishii, J.; Betel, D.; Alonso, A.; Mason, C.E.; Figueroa, M.E.; Melnick, A.M. Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J. Vis. Exp. 2015, e52246. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Wittbrodt, J. One for all—A highly efficient and versatile method for fluorescent immunostaining in fish embryos. PLoS ONE 2011, 6, e19713. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.; Fahimi, H.D. Immunocytochemical localization of albumin in the secretory apparatus of rat liver parenchymal cells. Proc. Natl. Acad. Sci. USA 1981, 78, 4970–4974. [Google Scholar] [CrossRef] [Green Version]
- Baratta, J.L.; Ngo, A.; Lopez, B.; Kasabwalla, N.; Longmuir, K.J.; Robertson, R.T. Cellular organization of normal mouse liver: A histological, quantitative immunocytochemical, and fine structural analysis. Histochem. Cell Biol. 2009, 131, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Shevelyov, Y.Y.; Ulianov, S.V. The Nuclear Lamina as an Organizer of Chromosome Architecture. Cells 2019, 8, 136. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.K.; Liu, R.Z.; Thisse, C.; Thisse, B.; Denovan-Wright, E.M.; Wright, J.M. Hierarchical subfunctionalization of fabp1a, fabp1b and fabp10 tissue-specific expression may account for retention of these duplicated genes in the zebrafish (Danio rerio) genome. FEBS J. 2006, 273, 3216–3229. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, A.B.; Thisse, C.; Thisse, B.; Wright, J.M. Differential tissue-specific distribution of transcripts for the duplicated fatty acid-binding protein 10 (fabp10) genes in embryos, larvae and adult zebrafish (Danio rerio). FEBS J. 2009, 276, 6787–6797. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef]
- Espada, J.; Esteller, M. Epigenetic control of nuclear architecture. Cell Mol. Life Sci. 2007, 64, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Salhab, A.; Nordstrom, K.; Gasparoni, G.; Kattler, K.; Ebert, P.; Ramirez, F.; Arrigoni, L.; Muller, F.; Polansky, J.K.; Cadenas, C.; et al. A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol. 2018, 19, 150. [Google Scholar] [CrossRef] [Green Version]
- Reznik, B.; Cincotta, S.A.; Jaszczak, R.G.; Mateo, L.J.; Shen, J.; Cao, M.; Baskin, L.; Ye, P.; An, W.; Laird, D.J. Heterogeneity of transposon expression and activation of the repressive network in human fetal germ cells. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grun, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Chen, Y.H.; Chen, H.L.; Chien, C.S.; Wu, S.H.; Ho, Y.T.; Yu, C.H.; Chang, M.H. Contribution of Mature Hepatocytes to Biliary Regeneration in Rats with Acute and Chronic Biliary Injury. PLoS ONE 2015, 10, e0134327. [Google Scholar] [CrossRef]
- Zhang, C.; Macchi, F.; Magnani, E.; Sadler, K.C. Chromatin states shaped by an epigenetic code confer regenerative potential to the mouse liver. Nat. Commun. 2021, 12, 4110. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, K.M.; Cohet, N.; Wu, Q.; Underwood, J.M.; Imbalzano, A.N.; Nickerson, J.A. Nuclear shape changes are induced by knockdown of the SWI/SNF ATPase BRG1 and are independent of cytoskeletal connections. PLoS ONE 2013, 8, e55628. [Google Scholar] [CrossRef] [Green Version]
- Espada, J.; Ballestar, E.; Santoro, R.; Fraga, M.F.; Villar-Garea, A.; Nemeth, A.; Lopez-Serra, L.; Ropero, S.; Aranda, A.; Orozco, H.; et al. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res. 2007, 35, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Gagnon-Kugler, T.; Langlois, F.; Stefanovsky, V.; Lessard, F.; Moss, T. Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase II transcription and disrupts ribosomal RNA processing. Mol. Cell 2009, 35, 414–425. [Google Scholar] [CrossRef]
- Adam, S.A.; Butin-Israeli, V.; Cleland, M.M.; Shimi, T.; Goldman, R.D. Disruption of lamin B1 and lamin B2 processing and localization by farnesyltransferase inhibitors. Nucleus 2013, 4, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derenzini, M.; Ploton, D. Interphase nucleolar organizer regions in cancer cells. Int. Rev. Exp. Pathol. 1991, 32, 149–192. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Trere, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Zabulica, M.; Srinivasan, R.C.; Vosough, M.; Hammarstedt, C.; Wu, T.; Gramignoli, R.; Ellis, E.; Kannisto, K.; Collin de l’Hortet, A.; Takeishi, K.; et al. Guide to the Assessment of Mature Liver Gene Expression in Stem Cell-Derived Hepatocytes. Stem. Cells Dev. 2019, 28, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, L.T.; Tan, A.K.Y.; Autio, M.I.; Goh, S.H.; Choo, S.H.; Lee, K.L.; Tan, J.; Pan, B.; Lee, J.J.H.; Lum, J.J.; et al. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep. 2018, 22, 2190–2205. [Google Scholar] [CrossRef] [Green Version]
- Gorkin, D.U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A.Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 2020, 583, 744–751. [Google Scholar] [CrossRef]
- Chen, T.; Oh, S.; Gregory, S.; Shen, X.; Diehl, A.M. Single-cell omics analysis reveals functional diversification of hepatocytes during liver regeneration. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Su, X.; Shi, Y.; Zou, X.; Lu, Z.N.; Xie, G.; Yang, J.Y.H.; Wu, C.C.; Cui, X.F.; He, K.Y.; Luo, Q.; et al. Single-cell RNA-Seq analysis reveals dynamic trajectories during mouse liver development. BMC Genome 2017, 18, 946. [Google Scholar] [CrossRef]
- Dong, J.; Hu, Y.; Fan, X.; Wu, X.; Mao, Y.; Hu, B.; Guo, H.; Wen, L.; Tang, F. Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018, 19, 31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Gu, C.; Yang, L.; Tang, F.; Gao, Y.Q. The sequence preference of DNA methylation variation in mammalians. PLoS ONE 2017, 12, e0186559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Nagrajan, H.K.; Yi, S.V. Fundamental diversity of human CpG islands at multiple biological levels. Epigenetics 2014, 9, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef]
- Chang, N.-C.; Rovira, Q.; Wells, J.N.; Feschotte, C.; Vaquerizas, J.M. A genomic portrait of zebrafish transposable elements and their spatiotemporal embryonic expression. bioRxiv 2021. [Google Scholar] [CrossRef]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Mazumder, A.; Roopa, T.; Kumar, A.; Iyer, K.V.; Ramdas, N.M.; Shivashankar, G.V. Prestressed nuclear organization in living cells. Methods Cell Biol. 2010, 98, 221–239. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Hampoelz, B.; Lecuit, T. Nuclear mechanics in differentiation and development. Curr. Opin. Cell Biol. 2011, 23, 668–675. [Google Scholar] [CrossRef]
- Schreiner, S.M.; Koo, P.K.; Zhao, Y.; Mochrie, S.G.; King, M.C. The tethering of chromatin to the nuclear envelope supports nuclear mechanics. Nat. Commun. 2015, 6, 7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendes, H.D.; Keyl, H.G. Distribution of DNA in heterochromatin and euchromatin of polytene nuclei of Drosophila hydei. Genetics 1967, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Politz, J.C.; Scalzo, D.; Groudine, M. Something silent this way forms: The functional organization of the repressive nuclear compartment. Annu Rev. Cell Dev. Biol. 2013, 29, 241–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Feinberg, A.P. Higher order chromatin organization in cancer. Semin. Cancer Biol. 2013, 23, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.; Cook, A.W.; Gough, R.E.; Schilling, M.; Olszok, N.A.; Brown, I.; Wang, L.; Aaron, J.; Martin-Fernandez, M.L.; Rehfeldt, F.; et al. DNA damage alters nuclear mechanics through chromatin reorganization. Nucleic Acids Res. 2021, 49, 340–353. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Marko, J.F. Chromatin’s physical properties shape the nucleus and its functions. Curr. Opin. Cell Biol. 2019, 58, 76–84. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Dahl, K.N.; Kahn, S.M.; Wilson, K.L.; Discher, D.E. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J. Cell Sci. 2004, 117, 4779–4786. [Google Scholar] [CrossRef] [Green Version]
- De Vos, W.H.; Houben, F.; Hoebe, R.A.; Hennekam, R.; van Engelen, B.; Manders, E.M.; Ramaekers, F.C.; Broers, J.L.; Van Oostveldt, P. Increased plasticity of the nuclear envelope and hypermobility of telomeres due to the loss of A-type lamins. Biochim. Biophys. Acta 2010, 1800, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, G.; Columbaro, M.; Mattioli, E.; Cenni, V.; Camozzi, D.; Wehnert, M.; Santi, S.; Riccio, M.; Del Coco, R.; Maraldi, N.M.; et al. Pre-Lamin A processing is linked to heterochromatin organization. J. Cell Biochem. 2007, 102, 1149–1159. [Google Scholar] [CrossRef]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Santi, S.; Maraldi, N.M.; D’Apice, M.R.; Novelli, G.; Riccio, M.; Squarzoni, S.; Foisner, R.; et al. Drugs affecting prelamin A processing: Effects on heterochromatin organization. Exp. Cell Res. 2008, 314, 453–462. [Google Scholar] [CrossRef]
- Broers, J.L.; Hutchison, C.J.; Ramaekers, F.C. Laminopathies. J. Pathol. 2004, 204, 478–488. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Burla, R.; La Torre, M.; Merigliano, C.; Verni, F.; Saggio, I. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 2018, 9, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic Reorganization of Lamin-Associated Domains in Cardiac Myocytes Is Associated With Differential Gene Expression and DNA Methylation in Human Dilated Cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, E.; Imamura, S.; Qi, J.; Toure, J.; Valdez, D.M., Jr.; Carr, C.E.; Hanai, J.; Kishi, S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS ONE 2011, 6, e17688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiello, I.; Biggiogera, M. Ultrastructural localization of 5-methylcytosine on DNA and RNA. Cell Mol. Life Sci. 2017, 74, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Verdun, D. The nucleolus: A model for the organization of nuclear functions. Histochem. Cell Biol. 2006, 126, 135–148. [Google Scholar] [CrossRef]
- Hernandez-Verdun, D. Assembly and disassembly of the nucleolus during the cell cycle. Nucleus 2011, 2, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Helpap, B. Observations on the number, size and localization of nucleoli in hyperplastic and neoplastic prostatic disease. Histopathology 1988, 13, 203–211. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 1983, 111, 47–54. [Google Scholar] [CrossRef]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Ashraf, W.; Ibrahim, A.; Alhosin, M.; Zaayter, L.; Ouararhni, K.; Papin, C.; Ahmad, T.; Hamiche, A.; Mely, Y.; Bronner, C.; et al. The epigenetic integrator UHRF1: On the road to become a universal biomarker for cancer. Oncotarget 2017, 8, 51946–51962. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Preston, R.; Santella, R.M.; Blanco, A.; Desai, M.; Berdasco, M.; Fraga, M. Global DNA hypomethylation in liver cancer cases and controls: A phase I preclinical biomarker development study. Epigenetics 2007, 2, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Conrad, R.; Castelino-Prabhu, S.; Cobb, C.; Raza, A. Cytopathologic diagnosis of liver mass lesions. J. Gastrointest. Oncol. 2013, 4, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Z.D. Neoplasms of the liver. Mod. Pathol. 2007, 20 (Suppl. 1), S49–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, B.C.; Waldburger, N.; Schlamp, K.; Jager, D.; Weiss, K.H.; Schulze-Bergkamen, H.; Schirmacher, P.; Springfeld, C. Liver cancers with stem/progenitor-cell features—A rare chemotherapy-sensitive malignancy. Oncotarget 2017, 8, 59991–59998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Li, P.; Fang, L.; Zhu, H.; Xu, L.; Cheng, H.; Zhang, J.; Li, F.; Feng, Y.; Li, Y.; et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016, 2, 16007. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Tanaka, Y.; Su, J.; Cakir, B.; Xiang, Y.; Patterson, B.; Ding, J.; Jung, Y.W.; Kim, J.H.; Hysolli, E.; et al. Uhrf1 regulates active transcriptional marks at bivalent domains in pluripotent stem cells through Setd1a. Nat. Commun. 2018, 9, 2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madakashira, B.P.; Zhang, C.; Macchi, F.; Magnani, E.; Sadler, K.C. Nuclear Organization during Hepatogenesis in Zebrafish Requires Uhrf1. Genes 2021, 12, 1081. https://doi.org/10.3390/genes12071081
Madakashira BP, Zhang C, Macchi F, Magnani E, Sadler KC. Nuclear Organization during Hepatogenesis in Zebrafish Requires Uhrf1. Genes. 2021; 12(7):1081. https://doi.org/10.3390/genes12071081
Chicago/Turabian StyleMadakashira, Bhavani P., Chi Zhang, Filippo Macchi, Elena Magnani, and Kirsten C. Sadler. 2021. "Nuclear Organization during Hepatogenesis in Zebrafish Requires Uhrf1" Genes 12, no. 7: 1081. https://doi.org/10.3390/genes12071081
APA StyleMadakashira, B. P., Zhang, C., Macchi, F., Magnani, E., & Sadler, K. C. (2021). Nuclear Organization during Hepatogenesis in Zebrafish Requires Uhrf1. Genes, 12(7), 1081. https://doi.org/10.3390/genes12071081