
The Importance of Being PI3K in the RAS Signaling Network


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
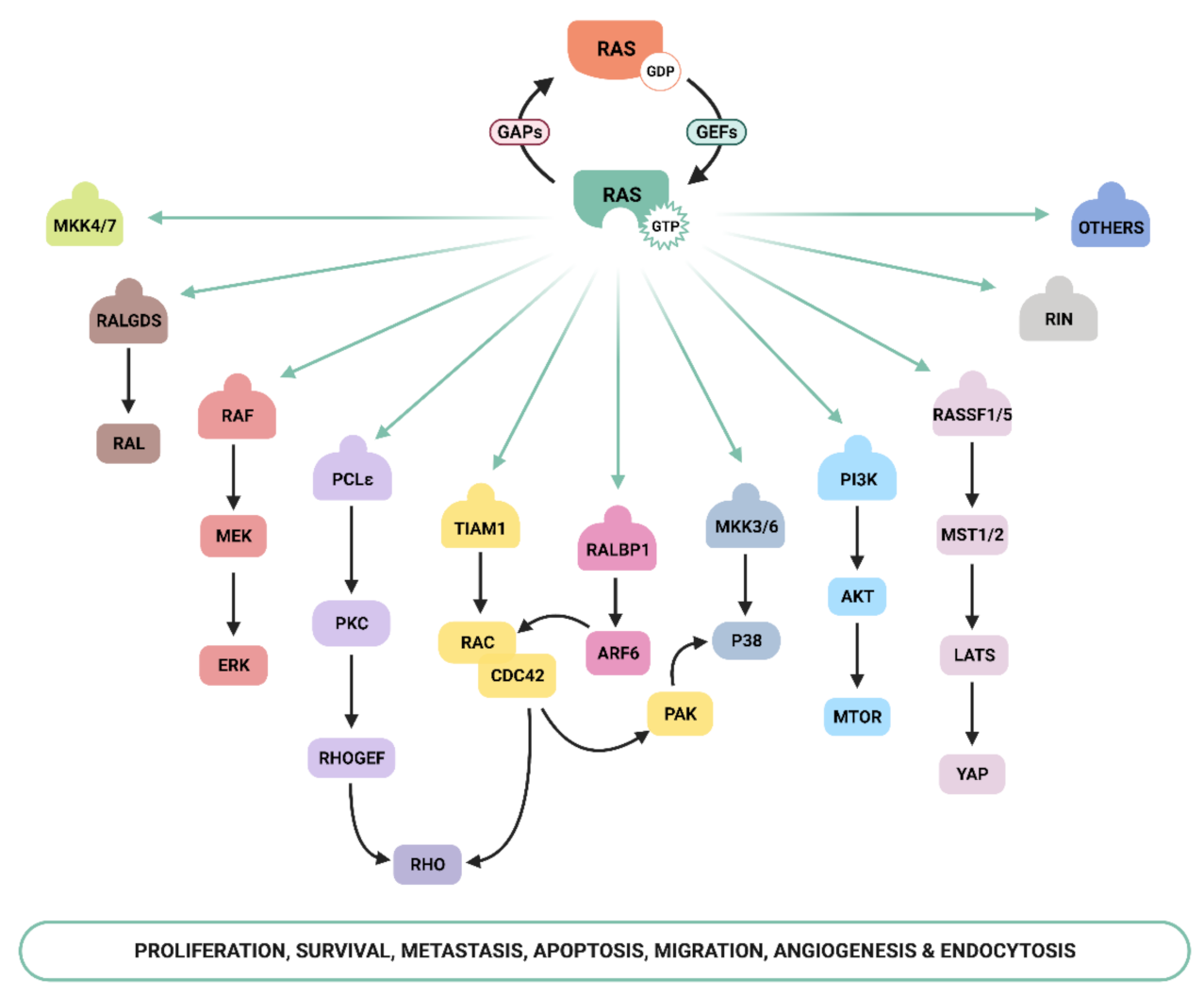
2. Ras Activation and Downstream Signaling
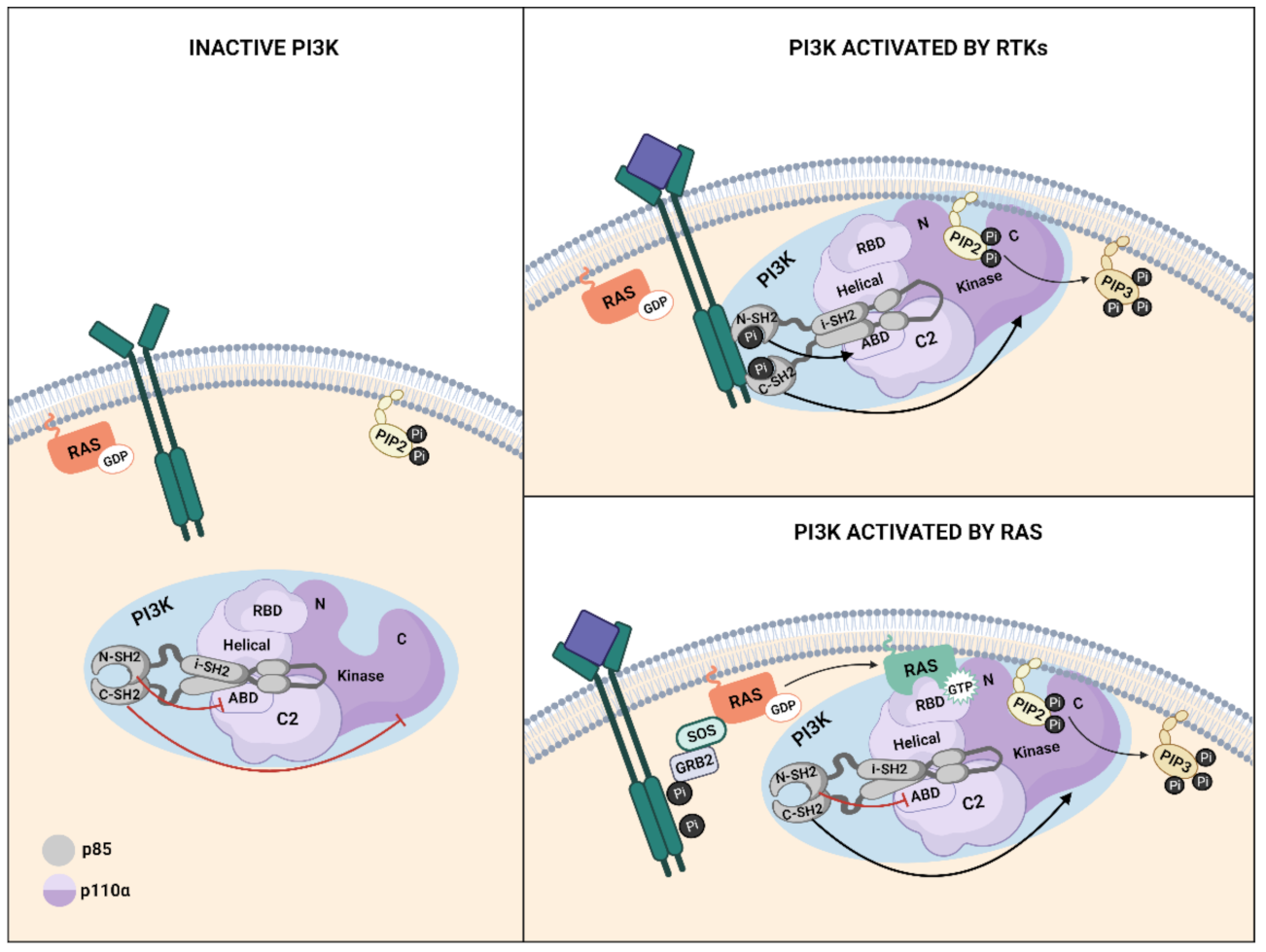
3. PI3K Family
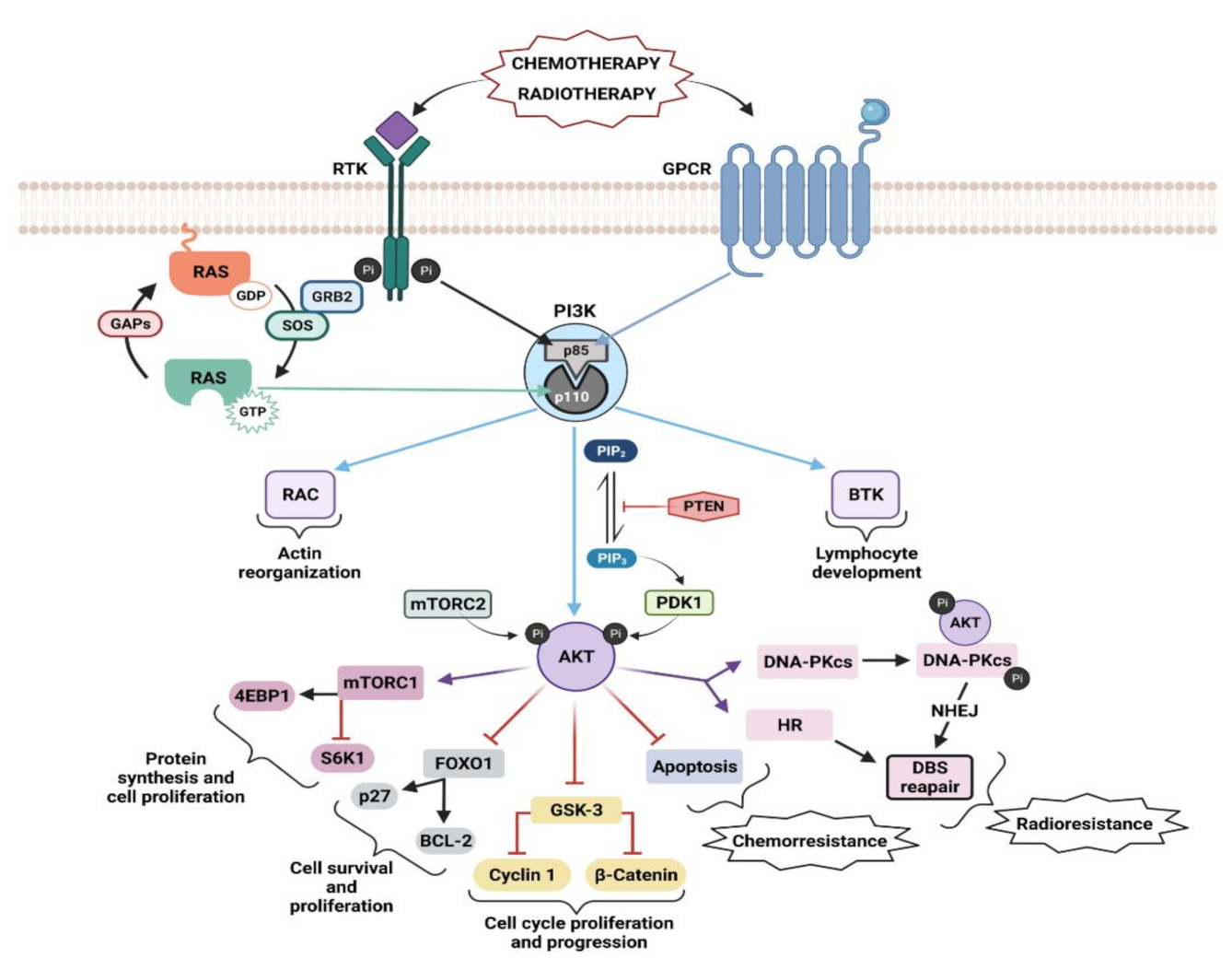
4. Ras–PI3K Interaction
4.1. Ras–PI3K Interaction in Cell Growth and Apoptosis
4.2. Ras–PI3K Signaling in the Immune System
5. Ras–PI3K Interaction in Cancer
5.1. Ras–PI3K Signaling in Migration and Metastasis
5.2. Ras–PI3K Signaling in Therapy Resistance
6. Ras–PI3K Signaling in the Tumor Microenvironment
6.1. Ras–PI3K Interaction in Angiogenesis
6.2. Ras–PI3K Interaction in Tumor Immune Infiltration
6.3. Ras–PI3K in CAFs and ECM Remodeling
7. Targeting Ras and PI3K in Cancer
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Martin, M.A.; Arenas, J.; Lucia, A.; Zugaza, J.L. Small GTPases of the Ras superfamily and glycogen phosphorylase regulation in T cells. Small GTPases 2021, 12, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krygowska, A.A.; Castellano, E. PI3K: A Crucial Piece in the RAS Signaling Puzzle. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.E.; Rubio, I.; Roose, J.P. Regulation of ras exchange factors and cellular localization of ras activation by lipid messengers in T cells. Front. Immunol. 2013, 4, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Mattos, C. The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb. Perspect. Med. 2018, 8, a031427. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; Wink, G.; Mattos, C. Transformation efficiency of Q61 Ras mutants linked to structural features of the switch regions. Structure 2007, 15, 1618–1629. [Google Scholar] [CrossRef] [Green Version]
- Henis, Y.I.; Hancock, J.F.; Prior, I.A. Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 2009, 26, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the und ruggable drugged? Nat. Rev. Drug. Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Czernilofsky, A.P.; Keri, G.; Smigelskaite, J.; Sill, H.; Troppmair, J. Signaling Through RAS-RAF-MEK-ERK: From Basics to Bedside. Curr. Med. Chem. 2007, 14, 601–623. [Google Scholar] [CrossRef]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenonos, K.; Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J. Int.Gastrointest. Oncol. 2013, 5, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fray, M.J.; Waterfield, M.D.; Donward, J. Phospatidylinositositol-3-OH kinase as a target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal Structure and Functional Analysisof Ras Binding to Its EffectorPhosphoinositide 3-Kinase. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Rebhun, J.F.; Chen, H.; Quilliam, L.A. Identification and Characterization of a New Family of Guanine Nucleotide Exchange Factors for the Ras-related GTPase Ral. J. Biol. Chem. 2000, 275, 13406–13410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent proteinkinase which phosphorylates and activates protein kinase Ba. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Zinatizadeh, M.R.; Miri, S.R.; Zarandi, P.K.; Chalbatani, G.M.; Raposo, C.; Mirzaei, H.R.; Akbari, M.E.; Mahmoodzadeh, H. The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in cancer metastasis. Genes. Dis. 2021, 8, 48–60. [Google Scholar] [CrossRef]
- Kiel, C.; Foglierini, M.; Kuemmerer, N.; Beltrao, P.; Serrano, L. A genome-wide Ras-effector interaction network. J. Mol. Biol. 2007, 370, 1020–1032. [Google Scholar] [CrossRef]
- Ibanez Gaspar, V.; Catozzi, S.; Ternet, C.; Luthert, P.J.; Kiel, C. Analysis of Ras-effector interaction competition in large intestine and colorectal cancer context. Small GTPases 2021, 12, 209–225. [Google Scholar] [CrossRef] [Green Version]
- Erijman, A.; Shifman, J.M. RAS/Effector Interactions from Structural and Biophysical Perspective. Mini. Rev. Med. Chem. 2016, 16, 370–375. [Google Scholar] [CrossRef]
- Catozzi, S.; Halasz, M.; Kiel, C. Predicted ‘wiring landscape’ of Ras-effector interactions in 29 human tissues. NPJ. Syst. Biol. Appl. 2021, 7, 10. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Foster, F.M.; Traer, C.J.; Abraham, S.M.; Fry, M.J. The phosphoinositide (PI) 3-kinase family. J. Cell Sci. 2003, 116, 3037–3040. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.A.; Dornan, G.L.; Hessenberger, M.; Haucke, V. The molecular mechanisms mediating class II PI 3-kinase function in cell physiology. FEBS J. 2021. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhou, W.; Chen, J.; Wei, W. Upstream regulators of phosphoinositide 3-kinase and their role in diseases. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R. The mechanism of PI3Kalpha activation at the atomic level. Chem. Sci. 2019, 10, 3671–3680. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segaliny, A.I.; Tellez-Gabriel, M.; Heymann, M.F.; Heymann, D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J. Bone Oncol. 2015, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Auger, K.R.; Chatterjee, S.; Terrence, R.; Burke, J.; Shoelson, S.E. Inhibition of SH2 Domain/Phosphoprotein Association by a Nonhydrolyzable Phosphonopeptide. Bio. Chem. 1992, 31, 9865–9870. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in Signal Transduction: From Phosphotyrosine-SH2 Domain Interactions to Complex Cellular Systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinaseby fibroblast growth factor receptors ismediated by coordinated recruitmentof multiple docking proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, C.L.; Auger, K.R.; Chanudhuri, M.; Yoakim, M.; Schaffhausen, B.; Shoelson, S.; Cantley, L.C. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 1993, 268, 9478–9483. [Google Scholar] [CrossRef]
- Brock, C.; Schaefer, M.; Reusch, H.P.; Czupalla, C.; Michalke, M.; Spicher, K.; Schultz, G.; Nurnberg, B. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J. Cell Bio Biol. 2003, 160, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes. Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, C.W.; Morgan, M.A.; Bergmann, L. Targeting the Ras signaling pathway: A rational, mechanism-based treatment for hematologic malignancies? Blood 2000, 96, 1655–1669. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulationof Akt/PKB by theRictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Haar, E.V.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples SurvivalSignals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- She, Q.B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruta, F.; Masuyama, N.; Gotoh, Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 2002, 277, 14040–14047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinaseyAkt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [Green Version]
- Madhunapantula, S.V.; Mosca, P.J.; Robertson, G.P. The Akt signaling pathway: An emerging therapeutic target in malignant melanoma. Cancer Biol. Ther. 2011, 12, 1032–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, G.S.; Lin, C.H.; Yang, Y.L.; Wu, M.S.; Chen, B.C. Ghrelin induces colon cancer cell proliferation through the GHS-R, Ras, PI3K, Akt, and mTOR signaling pathways. Eur. J. Pharm. 2016, 776, 124–131. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Middendorp, S.; Dingjan, G.M.; Hendriks, R.W. Impaired precursor B cell differentiation in Bruton’s tyrosine kinase-deficient mice. J. Immunol. 2002, 168, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Middendorp, S.; Dingjan, G.M.; Maas, A.; Dahlenborg, K.; Hendriks, R.W. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol. 2003, 171, 5988–5996. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.T.; Berg, L.J. New insights into the regulation and functions of Tec family tyrosine kinases in the immune system. Curr. Opin. Immunol. 2002, 14, 331–340. [Google Scholar] [CrossRef]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarstrom, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Ann. Annu. Rev. Immunol. 2004, 22, 563–598. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug. Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef] [PubMed]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Viernes, D.R.; Choi, L.B.; Kerr, W.G.; Chisholm, J.D. Discovery and development of small molecule SHIP phosphatase modulators. Med. Res. Rev. 2014, 34, 795–824. [Google Scholar] [CrossRef] [PubMed]
- Miletic, A.V.; Anzelon-Mills, A.N.; Mills, D.M.; Omori, S.A.; Pedersen, I.M.; Shin, D.M.; Ravetch, J.V.; Bolland, S.; Morse, H.C., 3rd; Rickert, R.C. Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J. Exp. Med. 2010, 207, 2407–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the "undruggable". Bio. Chim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denley, A.; Kang, S.; Karst, U.; Vogt, P.K. Oncogenic signaling of class I PI3K isoforms. Oncogene 2008, 27, 2561–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1231. [Google Scholar] [CrossRef]
- Buckles, T.C.; Ziemba, B.P.; Masson, G.R.; Williams, R.L.; Falke, J.J. Single-Molecule Study Reveals How Receptor and Ras Synergistically Activate PI3Kalpha and PIP3 Signaling. Biophys. J. 2017, 113, 2396–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate.p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Geering, B.; Cutillas, P.R.; Vanhaesebroeck, B. Regulation of class IA PI3Ks: Is there a role for monomeric PI3K subunits? Biochem. Soc. Trans. 2007, 35, 199–203. [Google Scholar] [CrossRef]
- Burke, J.E.; Perisic, O.; Masson, G.R.; Vadas, O.; Williams, R.L. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc. Natl. Acad. Sci. USA 2012, 109, 15259–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85p110 Phosphatidylinositol 3′-Kinase Stabilization and Inhibition of the p110α Catalytic Subunit. Mol. Cell Biol. 1998, 18, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.; Schaffhausen, B.; Liu, Y.; Günther, U.L. NMR Structure of the N-SH2 of the p85 Subunit of Phosphoinositide 3-Kinase Complexed to a Doubly Phosphorylated Peptide Reveals a Second Phosphotyrosine Binding Site †. Biochemical 2000, 39, 15860–15869. [Google Scholar] [CrossRef]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kalpha by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.-L.; Wang, J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3085–3096. [Google Scholar]
- Rodriguez-Viciana, P.; Warne, P.H.; Khwaja, A.; Marte, B.M.; Pappin, D.; Das, P.; Waterfield, M.D.; Ridley, A.; Downward, J. Role of Phosphoinositide 3-OH Kinase in Cell Transformation and Control of the Actin Cytoskeleton by Ras. Cell 1997, 87, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Suire, S.; Condliffe, A.M.; Ferguson, G.J.; Ellson, C.D.; Guillou, H.; Davidson, K.; Welch, H.; Coadwell, J.; Turner, M.; Chilvers, E.R.; et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat. Cell Biol. 2006, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Sjolander, A.; Yamamoto, K.; Huber, B.E.; Lapetina, E.G. Association of p21ras with phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 7908–7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, H.; Shao, J.; DuBois, R.N. Akt/PKB Activity Is Required for Ha-Ras-mediated Transformation of Intestinal Epithelial Cells. J. Biol. Chem. 2001, 276, 14498–14504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.A.; Park, M.T.; Kang, C.M.; Um, H.D.; Bae, S.; Lee, K.H.; Kim, T.H.; Kim, J.H.; Cho, C.K.; Lee, Y.S.; et al. Opposite effects of Ha-Ras and Ki-Ras on radiation-induced apoptosis via differential activation of PI3K/Akt and Rac/p38 mitogen-activated protein kinase signaling pathways. Oncogene 2004, 23, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritsman, K.; Yuzugullu, H.; Von, T.; Yan, H.; Clayton, L.; Fritsch, C.; Maira, S.M.; Hollingworth, G.; Choi, C.; Khandan, T.; et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J. Clin. Investig. 2014, 124, 1794–1809. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Iwanaga, K.; Raso, M.G.; Wislez, M.; Hanna, A.E.; Wieder, E.D.; Molldrem, J.J.; Wistuba, I.I.; Powis, G.; Demayo, F.J.; et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS ONE 2008, 3, e2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Han, Y.K.; Kim, J.Y.; Lee, E.G.; Lee, H.W.; Lee, G.M. Effect of constitutively active Ras overexpression on cell growth in recombinant Chinese hamster ovary cells. Biotechnol. Prog. 2011, 27, 577–580. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Gilbert, E.U.C.; Coffert, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef]
- Orme, M.H.; Alrubaie, S.; Bradley, G.L.; Walker, C.D.; Leevers, S.J. Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat. Cell Biol. 2006, 8, 1298–1302. [Google Scholar] [CrossRef]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes. Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Chun-Jen Lin, C.; Stern, M. Ras-dependent and Ras-independent effects of PI3K in Drosophila motor neurons. Genes. Brain Behav. 2012, 11, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Murillo, M.M.; Rana, S.; Spencer-Dene, B.; Nye, E.; Stamp, G.; Downward, J. Disruption of the Interaction of RAS with PI 3-Kinase Induces Regression of EGFR-Mutant-Driven Lung Cancer. Cell Rep. 2018, 25, 3545–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, A.; Becattini, B.; Mazzoli, A.; Bleve, A.; Radici, L.; Maxvall, I.; Sopasakis, V.R.; Molinaro, A.; Backhed, F.; Solinas, G. Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kalpha and PI3Kbeta Activities and Is Promoted by RAS. Cell Metab. 2019, 29, 1400–1409.e1405. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Klinghoffer, R.; Prestwich, G.D.; Toker, A.; Kazlauskas, A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Curr. Biol. 1999, 9, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Whiteford, C.C.; Best, C.; Kazlauskas, A.; Ulug, E.T. D-3 phosphoinositide metabolism in cells treated with platelet-derived growth factor. Bio. Chem. J. 1996, 319, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auger, K.R.; Serunian, L.A.; Soltoff, S.P.; Libby, P.; Cantley, L.C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Gire, V.; Marshall, C.; Wynford-Thomas, D. PI-3-kinase is an essential anti-apoptotic effector in the proliferative response of primary human epithelial cells to mutant RAS. Oncogene 2000, 19, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Sethi, T.; Ginsberg, M.H.; Downward, J.; Hughes, P.E. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol. Biol. Cell 1999, 10, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Ruoslahti, E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 2005, 11, 1346–1350. [Google Scholar] [CrossRef]
- Li, F.; Sawada, J.; Komatsu, M. R-Ras-Akt axis induces endothelial lumenogenesis and regulates the patency of regenerating vasculature. Nat. Commun. 2017, 8, 1720. [Google Scholar] [CrossRef]
- Sawada, J.; Urakami, T.; Li, F.; Urakami, A.; Zhu, W.; Fukuda, M.; Li, D.Y.; Ruoslahti, E.; Komatsu, M. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 2012, 22, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, G.S.; Grundl, M.; Allen, J.S., 3rd; Matter, M.L. R-Ras interacts with filamin a to maintain endothelial barrier function. J. Cell Physiol. 2011, 226, 2287–2296. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.I.; Sato, T.; Nagamine, K.; Hori, T. The inhibition of Bax activation-induced apoptosis by RasGRP2 via R-Ras-PI3K-Akt signaling pathway in the endothelial cells. Sci. Rep. 2019, 9, 16717. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Morishita, R.; Yamamoto, K.; Yoshimura, S.I.; Taniyama, Y.; Aoki, M.; Matsubara, H.; Kim, S.; Kaneda, Y.; Ogihara, T. Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes 2001, 50, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Jiang, Y.; Fu, T.; Hao, Y.; Zhu, X.; Lu, Y. Naringin inhibits lipopolysaccharide-induced damage in human umbilical vein endothelial cells via attenuation of inflammation, apoptosis and MAPK pathways. Cytotechnology 2016, 68, 1473–1487. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.A.; Rawlings, D.; Reynolds, H.; Vigorito, E.; et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Bio. Biophys. Acta. Mol. Cell Biol. Lipids 2015, 1851, 882–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banham-Hall, E.; Clatworthy, M.R.; Okkenhaug, K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Perez de Castro, I.; Diaz, R.; Malumbres, M.; Hernandez, M.I.; Jagirdar, J.; Jimenez, M.; Ahn, D.; Pellicer, A. Mice deficient for N-ras: Impaired antiviral immune response and T-cell function. Cancer Res. 2003, 63, 1615–1622. [Google Scholar]
- Kurig, B.; Shymanets, A.; Bohnacker, T.; Prajwal; Brock, C.; Ahmadian, M.R.; Schaefer, M.; Gohla, A.; Harteneck, C.; Wymann, M.P.; et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc. Natl. Acad. Sci. USA 2009, 106, 20312–20317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, P.; Dorner, M.B.; Schaefer, M. Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006, 281, 9977–9986. [Google Scholar] [CrossRef] [Green Version]
- Nürnberg, B.; Beer-Hammer, S. Function, Regulation and Biological Roles of PI3Kγ Variants. Biol. Mol. 2019, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Shymanets, A.; Prajwal; Bucher, K.; Beer-Hammer, S.; Harteneck, C.; Nurnberg, B. p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J. Biol. Chem. 2013, 288, 31059–31068. [Google Scholar] [CrossRef] [Green Version]
- Rynkiewicz, N.K.; Anderson, K.E.; Suire, S.; Collins, D.M.; Karanasios, E.; Vadas, O.; Williams, R.; Oxley, D.; Clark, J.; Stephens, L.R.; et al. Gbetagamma is a direct regulator of endogenous p101/p110gamma and p84/p110gamma PI3Kgamma complexes in mouse neutrophils. Sci. Signal 2020, 13, 1289. [Google Scholar] [CrossRef] [PubMed]
- Deladeriere, A.; Gambardella, L.; Pan, D.; Anderson, K.E.; Hawkins, P.T.; Stephens, L.R. The regulatory subunits of PI3Kgamma control distinct neutrophil responses. Sci. Signal 2015, 8, ra8. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.R.; Gogvadze, E.; Xavier, A.R.; Bohnacker, T.; Voelzmann, J.; Wymann, M.P. PI3Kgamma Regulatory Protein p84 Determines Mast Cell Sensitivity to Ras Inhibition-Moving Towards Cell Specific PI3K Targeting? Front. Immunol. 2020, 11, 585070. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a New Gβγ-Activated Regulatory Subunit of the Type IB Phosphoinositide 3-Kinase p110γ. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Suire, S.; Baltanas, F.C.; Segonds-Pichon, A.; Davidson, K.; Santos, E.; Hawkins, P.T.; Stephens, L.R. Frontline Science: TNF-alpha and GM-CSF1 priming augments the role of SOS1/2 in driving activation of Ras, PI3K-gamma, and neutrophil proinflammatory responses. J. Leukoc. Biol. 2019, 106, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suire, S.; Lecureuil, C.; Anderson, K.E.; Damoulakis, G.; Niewczas, I.; Davidson, K.; Guillou, H.; Pan, D.; Jonathan, C.; Phillip, T.H.; et al. GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J. 2012, 31, 3118–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffargue, M.; Calvez, R.; Finan, P.; Trifilieff, A.; Barbier, M.; Altruda, F.; Hirsch, E.; Wymann, M.P. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 2002, 16, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, A.; Ayesh, H.S.K.; Halawani, H. PIK3CA Gene Mutations in Solid Malignancies: Association with Clinicopathological Parameters and Prognosis. Cancers 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.E.; Williams, R.L. Synergy in activating class I PI3Ks. Trends Biochem. Sci. 2015, 40, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Miled, N.; Yan, Y.; Perisic, W.-C.H.O.; Inbar, M.Z.Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of Two Classes of Cancer Mutations in the Phosphoinositide 3-Kinase Catalytic Subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, A.; Bussaglia, E.; Pallares, J.; Dolcet, X.; Llobet, D.; Encinas, M.; Llecha, N.; Palacios, J.; Prat, J.; Matias-Guiu, X. PIK3CA gene mutations in endometrial carcinoma: Correlation with PTEN and K-RAS alterations. Hum. Pathol. 2006, 37, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lee, J.E.; Lee, S.S.; Kim, C.; Lee, S.J.; Jang, W.S.; Park, S. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int. J. Cancer 2013, 133, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, H.; Pan, Y.; Wang, R.; Li, Y.; Shen, L.; Yu, Y.; Li, H.; Cai, D.; Sun, Y.; et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE 2014, 9, e88291. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef]
- Leone, V.; di Palma, A.; Ricchi, P.; Acquaviva, F.; Giannouli, M.; Di Prisco, A.M.; Iuliano, F.; Acquaviva, A.M. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G673–G681. [Google Scholar] [CrossRef]
- Zhan, H.; Bhattacharya, S.; Cai, H.; Iglesias, P.A.; Huang, C.H.; Devreotes, P.N. An Excitable Ras/PI3K/ERK Signaling Network Controls Migration and Oncogenic Transformation in Epithelial Cells. Dev. Cell 2020, 54, 608–623. [Google Scholar] [CrossRef]
- Jiang, K.; Sun, J.; Cheng, J.; Djeu, J.Y.; Wei, S.; Sebti, S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol. Cell Biol. 2004, 24, 5565–5576. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Sheridan, C.; Thin, M.Z.; Nye, E.; Spencer-Dene, B.; Diefenbacher, M.E.; Moore, C.; Kumar, M.S.; Murillo, M.M.; Gronroos, E.; et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell 2013, 24, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Counter, C.M. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell 2005, 8, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebi, H.; Corcoran, R.B.; Singh, A.; Chen, Z.; Song, Y.; Lifshits, E.; Ryan, D.P.; Meyerhardt, J.A.; Benes, C.; Settleman, J.; et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Investig. 2011, 121, 4311–4321. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schonhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes. Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, D.W.; Watson, T.; Kung, H.F.; Curran, T. Microinjection of transforming ras protein induces c-fos expression. Mol. Cell Biol. 1987, 7, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Gutman, A.; Wasylyk, C.; Wasylyk, B. Cell-specific regulation of oncogene-responsive sequences of the c-fos promoter. Mol. Cell Biol. 1991, 11, 5381–5387. [Google Scholar] [CrossRef] [Green Version]
- Urich, M.; Senften, M.; Shaw, P.E.; Ballmer-Hofer, K. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene 1997, 14, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwick, J.K.; Cox, A.D.; Der, C.J.; Cobb, M.H.; Hibi, M.; Karin, M.; Brenner, D.A. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6030–6034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finco, T.S.; Westwick, J.K.; Norris, J.L.; Beg, A.A.; Der, C.J.; Baldwin, A.S., Jr. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J. Biol. Chem. 1997, 272, 24113–24116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Pellicer, A. RAS pathways to cell cycle control and cell transformation. Front. Biol. Sci. 1998, 3, d887–d912. [Google Scholar] [CrossRef]
- Filmus, J.; Robles, A.I.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar] [PubMed]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, J.T.; Coats, S.R.; Wang, Y.Z.; Pledger, W.J. Regulation of the cell cycle machinery by oncogenic ras. Oncogene 1996, 12, 127–134. [Google Scholar]
- Liu, J.J.; Chao, J.R.; Jiang, M.C.; Ng, S.Y.; Yen, J.J.; Yang-Yen, H.F. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell Biol. 1995, 15, 3654–3663. [Google Scholar] [CrossRef] [Green Version]
- Gille, H.; Downward, J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwick, J.K.; Lambert, Q.T.; Clark, G.J.; Symons, M.; Van Aelst, L.; Pestell, R.G.; Der, C.J. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol. Cell Biol. 1997, 17, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol. Cell Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes. Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Ottmann, O.G.; Deininger, M.; Hochhaus, A. Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica 2014, 99, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Janas, M.L.; Turner, M. Interaction of Ras with P110γ Is Required for Thymic β-Selection in the Mouse. J. Immunol. 2011, 187, 4667–4675. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Alam, H.; Tang, M.; Maitituoheti, M.; Dhar, S.S.; Kumar, M.; Han, C.Y.; Ambati, C.R.; Amin, S.B.; Gu, B.; Chen, T.Y.; et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 2020, 37, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Kim, K.H.; Sung, H.; Paraiso, K.H.; Dannenberg, J.H.; Bosenberg, M.; Chin, L.; Kim, M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010, 29, 6222–6232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Blenis, J. A nexus for cellular homeostasis: The interplay between metabolic and signal transduction pathways. Curr. Opin. Biotechnol. 2015, 34, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.G.; Sanders, A.J.; Katoh, M.; Ungefroren, H.; Gieseler, F.; Prince, M.; Thompson, S.K.; Zollo, M.; Spano, D.; Dhawan, P.; et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35, S244–S275. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Chiu, H.C.; Li, C.J.; Yiang, G.T.; Tsai, A.P.; Wu, M.Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef]
- Qu, T.; Zhao, Y.; Chen, Y.; Jin, S.; Fang, Y.; Jin, X.; Sun, L.; Ma, Y. Down-regulated MAC30 expression inhibits breast cancer cell invasion and EMT by suppressing Wnt/beta-catenin and PI3K/Akt signaling pathways. Int. J. Clin. Exp. Pathol. 2019, 12, 1888–1896. [Google Scholar]
- Xu, E.; Xia, X.; Jiang, C.; Li, Z.; Yang, Z.; Zheng, C.; Wang, X.; Du, S.; Miao, J.; Wang, F.; et al. GPER1 Silencing Suppresses the Proliferation, Migration, and Invasion of Gastric Cancer Cells by Inhibiting PI3K/AKT-Mediated EMT. Front. Cell Dev. Biol. 2020, 8, 591239. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Liang, C.; Li, Z.; An, D.; Ren, S.; Yue, C.; Wu, J. DcR3 induces proliferation, migration, invasion, and EMT in gastric cancer cells via the PI3K/AKT/GSK-3beta/beta-catenin signaling pathway. Onco Targets Ther. 2018, 11, 4177–4187. [Google Scholar] [CrossRef] [Green Version]
- Andreolas, C.; Kalogeropoulou, M.; Voulgari, A.; Pintzas, A. Fra-1 regulates vimentin during Ha-RAS-induced epithelial mesenchymal transition in human colon carcinoma cells. Int. J. Cancer 2008, 122, 1745–1756. [Google Scholar] [CrossRef]
- Li, B.; Xu, W.W.; Lam, A.K.Y.; Wang, Y.; Hu, H.F.; Guan, X.Y.; Qin, Y.R.; Saremi, N.; Tsao, S.W.; He, Q.Y.; et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 2017, 8, 38755–38766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.N.; Herrera, B.; Mikula, M.; Proell, V.; Fuchs, E.; Gotzmann, J.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis. Carcinogenesis 2005, 26, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre-Perona, A.; Riesco-Eizaguirre, G.; Zaballos, M.A.; Santisteban, P. beta-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget 2016, 7, 49435–49449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annibaldi, A.; Dousse, A.; Martin, S.; Tazi, J.; Widmann, C. Revisiting G3BP1 as a RasGAP binding protein: Sensitization of tumor cells to chemotherapy by the RasGAP 317-326 sequence does not involve G3BP1. PLoS ONE 2011, 6, e29024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitard, E.; Parker, F.; Millon, R.; Abecassis, J.; Tocque, B. G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett. 2001, 162, 213–221. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.H.; He, H.W.; Zhang, C.X.; Yu, D.K.; Shao, R.G. Downregulation of G3BPs inhibits the growth, migration and invasion of human lung carcinoma H1299 cells by suppressing the Src/FAK-associated signaling pathway. Cancer Gene Ther. 2013, 20, 622–629. [Google Scholar] [CrossRef]
- Zhang, L.N.; Zhao, L.; Yan, X.L.; Huang, Y.H. Loss of G3BP1 suppresses proliferation, migration, and invasion of esophageal cancer cells via Wnt/beta-catenin and PI3K/AKT signaling pathways. J. Cell Physiol. 2019, 234, 20469–20484. [Google Scholar] [CrossRef]
- Janda, E.; Litos, G.; Grunert, S.; Downward, J.; Beug, H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene 2002, 21, 5148–5159. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Giehl, K. Oncogenic Ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef]
- Grunert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; Borel Rinkes, I.H.; Voest, E.E.; Kranenburg, O. Control of colorectal metastasis formation by K-Ras. Bio.Chim. Biophys. Acta 2005, 1756, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005, 65, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- McCormick, B.; Chu, J.Y.; Vermeren, S. Cross-talk between Rho GTPases and PI3K in the neutrophil. Small GTPases 2019, 10, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Soriano, O.; Alcón-Pérez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819. [Google Scholar] [CrossRef]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [Green Version]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.E.; van Delft, S.; ten Klooster, J.P.; Reid, T.; van der Kammen, R.A.; Michiels, F.; Collard, J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 1998, 143, 1385–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liliental, J.; Moon, S.Y.; Lesche, R.; Mamillapalli, R.; Li, D.; Zheng, Y.; Sun, H.; Wu, H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 2000, 10, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaduwal, S.; Jeong, W.J.; Park, J.C.; Lee, K.H.; Lee, Y.M.; Jeon, S.H.; Lim, Y.B.; Min do, S.; Choi, K.Y. Sur8/Shoc2 promotes cell motility and metastasis through activation of Ras-PI3K signaling. Oncotarget 2015, 6, 33091–33105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, S.; Abdellatef, S.; Haddad, M.A.; Fakhoury, I.; El-Sibai, M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. [Google Scholar] [CrossRef] [Green Version]
- Jebri, I.; Tsujita, K.; Fujita, Y.; Itoh, T. Non-cell-autonomous migration of RasV12-transformed cells towards the basal side of surrounding normal cells. Biochem. Biophys. Res. Commun. 2021, 543, 15–22. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hung, M.C. Induction of Akt activity by chemotherapy confers acquired resistance. J. Formos. Med. Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Harari, P.M.; Wheeler, D.L.; Toulany, M. Targeting AKT/PKB to improve treatment outcomes for solid tumors. Mutat. Res. 2020, 819–820, 111690. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The Ras radiation resistance pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar] [PubMed]
- Toulany, M.; Dittmann, K.; Baumann, M.; Rodemann, H.P. Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS. Radiother Oncol. 2005, 74, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; Bae, S.S.; Fernandes, A.; Wu, J.; Muschel, R.J.; McKenna, W.G.; Birnbaum, M.J.; Bernhard, E.J. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 2005, 65, 7902–7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.Y.; Hu, S.L.; Xu, X.Y.; Wang, R.Z.; Yu, H.Y.; Xu, J.Y.; Chen, L.; Dong, G.L. Nimotuzumab enhances the radiosensitivity of cancer cells in vitro by inhibiting radiation-induced DNA damage repair. PLoS ONE 2013, 8, e70727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Kim, T.J.; Lee, J.W.; Song, S.Y.; Choi, J.J.; Choi, C.H.; Kim, B.G.; Lee, J.H.; Bae, D.S. Increased expression of pAKT is associated with radiation resistance in cervical cancer. Br. J. Cancer 2006, 94, 1678–1682. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [Green Version]
- Minjgee, M.; Toulany, M.; Kehlbach, R.; Giehl, K.; Rodemann, H.P. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int. J. Radiatoncol. Biol. 2011, 81, 1506–1514. [Google Scholar] [CrossRef]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 42700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, F.M.; Prior, I.A.; MacEwan, D.J. The importance of Ras in drug resistance in cancer. Br. J. Pharm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.J.; Ryu, Y.K.; Kim, S.Y.; Wu, H.G.; Kim, J.S.; Kim, I.H.; Kim, I.A. Targeting epidermal growth factor receptor-associated signaling pathways in non-small cell lung cancer cells: Implication in radiation response. Mol. Cancer Res. 2010, 8, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef] [PubMed]
- Weykamp, F.; Seidensaal, K.; Rieken, S.; Green, K.; Mende, S.; Zaoui, K.; Freier, K.; Adeberg, S.; Debus, J.; Welte, S.E. Age-dependent hemato- and nephrotoxicity in patients with head and neck cancer receiving chemoradiotherapy with weekly cisplatin. Strahlenther. Onkol. 2020, 196, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Caiola, E.; Salles, D.; Frapolli, R.; Lupi, M.; Rotella, G.; Ronchi, A.; Garassino, M.C.; Mattschas, N.; Colavecchio, S.; Broggini, M.; et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget 2015, 6, 30072–30087. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 2014, 20, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Garassino, M.C.; Marabese, M.; Rusconi, P.; Rulli, E.; Martelli, O.; Farina, G.; Scanni, A.; Broggini, M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 2011, 22, 235–237. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Luo, H.; Luo, D.; Yang, H.; Zhou, X. Src homology phosphotyrosyl phosphatase 2 mediates cisplatin-related drug resistance by inhibiting apoptosis and activating the Ras/PI3K/Akt1/survivin pathway in lung cancer cells. Oncol. Rep. 2018, 39, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, Y.N.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010, 70, 8736–8747. [Google Scholar] [CrossRef] [Green Version]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, P.P.; Cardone, C.; Martini, G.; Ciardiello, D.; Belli, V.; Matrone, N.; Barra, G.; Napolitano, S.; Della Corte, C.; Turano, M.; et al. Receptor tyrosine kinase-dependent PI3K activation is an escape mechanism to vertical suppression of the EGFR/RAS/MAPK pathway in KRAS-mutated human colorectal cancer cell lines. J. Exp. Clin. Cancer Res. 2019, 38, 41. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Won, J.K.; Yang, H.W.; Shin, S.Y.; Lee, J.H.; Heo, W.D.; Cho, K.H. The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J. Mol. Cell Biol. 2012, 4, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Amgen overcomes historically undruggable target, with FDA nod for first KRAS inhibitor. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Hata, A.N.; Shaw, A.T. Resistance looms for KRAS(G12C) inhibitors. Nat. Med. 2020, 26, 169–170. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRAS (G12C) Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS (G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Massarelli, E.; Varella-Garcia, M.; Tang, X.; Xavier, A.C.; Ozburn, N.C.; Liu, D.D.; Bekele, B.N.; Herbst, R.S.; Wistuba, I.I. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin. Cancer Res. 2007, 13, 2890–2896. [Google Scholar] [CrossRef] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef]
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixido, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzon, M.; Codony-Servat, C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- Riess, C.; Shokraie, F.; Classen, C.F.; Kreikemeyer, B.; Fiedler, T.; Junghanss, C.; Maletzki, C. Arginine-Depleting Enzymes—An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol. Biochem. 2018, 51, 854–870. [Google Scholar] [CrossRef]
- Tsai, W.B.; Aiba, I.; Long, Y.; Lin, H.K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Gebbink, M.F.; Voest, E.E. Stimulation of angiogenesis by Ras proteins. Biochim. Biophys. Acta. 2004, 1654, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Okada, F.; Rak, J.W.; Croix, B.S.; Lieubeau, B.; Kaya, M.; Roncari, L.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Impact of oncogenes in tumor angiogenesis: Mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3609–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar] [PubMed]
- Zhang, X.; Gaspard, J.P.; Chung, D.C. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001, 61, 6050–6054. [Google Scholar] [PubMed]
- Jung, F.; Haendeler, J.; Hoffmann, J.; Reissner, A.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Hypoxic induction of the hypoxia-inducible factor is mediated via the adaptor protein Shc in endothelial cells. Circ. Res. 2002, 91, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3’-kinase/Akt signaling pathway. Cancer Res. 2001, 61, 7349–7355. [Google Scholar] [PubMed]
- Lee, E.; Yim, S.; Lee, S.K.; Park, H. Two transactivation domains of hypoxia-inducible factor-1alpha regulated by the MEK-1/p42/p44 MAPK pathway. Mol. Cells 2002, 14, 9–15. [Google Scholar]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Jo, W.S.; Duerr, E.M.; Gala, M.; Li, J.; Zhang, X.; Zimmer, M.A.; Iliopoulos, O.; Zukerberg, L.R.; Kohgo, Y.; et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 2005, 11, 992–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Pathak, P.S.; Fukumura, D. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol 3’-kinase signaling pathways contributes to expression of interleukin 8 in human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunaga, N.; Imai, H.; Shimizu, K.; Shames, D.S.; Kakegawa, S.; Girard, L.; Sato, M.; Kaira, K.; Ishizuka, T.; Gazdar, A.F.; et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int. J. Cancer 2012, 130, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Ancrile, B.B.; O’Hayer, K.M.; Counter, C.M. Oncogenic ras-induced expression of cytokines: A new target of anti-cancer therapeutics. Mol. Interv. 2008, 8, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Rak, J.; Mitsuhashi, Y.; Sheehan, C.; Tamir, A.; Viloria-Petit, A.; Filmus, J.; Mansour, S.J.; Ahn, N.G.; Kerbel, R.S. Oncogenes and tumor angiogenesis: Differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000, 60, 490–498. [Google Scholar] [PubMed]
- Watnick, R.S.; Cheng, Y.N.; Rangarajan, A.; Ince, T.A.; Weinberg, R.A. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell 2003, 3, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Mazure, N.M.; Chen, E.Y.; Laderoute, K.R.; Giaccia, A.J. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997, 90, 3322–3331. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Meng, Q.; Cao, Z.; Shi, X.; Jiang, B.H. Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J. Cell Physiol. 2006, 209, 56–66. [Google Scholar] [CrossRef]
- Borrello, M.G.; Degl’Innocenti, D.; Pierotti, M.A. Inflammation and cancer: The oncogene-driven connection. Cancer Lett. 2008, 267, 262–270. [Google Scholar] [CrossRef]
- Feng, Y.; Santoriello, C.; Mione, M.; Hurlstone, A.; Martin, P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: Parallels between tumor initiation and wound inflammation. PLoS Biol. 2010, 8, e1000562. [Google Scholar] [CrossRef] [Green Version]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Dormond, O.; Foletti, A.; Paroz, C.; Ruegg, C. NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat. Med. 2001, 7, 1041–1047. [Google Scholar] [CrossRef]
- Sui, W.; Zhang, Y.; Wang, Z.; Wang, Z.; Jia, Q.; Wu, L.; Zhang, W. Antitumor effect of a selective COX-2 inhibitor, celecoxib, may be attributed to angiogenesis inhibition through modulating the PTEN/PI3K/Akt/HIF-1 pathway in an H(2)(2) murine hepatocarcinoma model. Oncol. Rep. 2014, 31, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Jia, Q.; Song, H.Y.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J. Biol. Chem. 1995, 270, 6729–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Choi, H.S.; Matsui, A.; Benes, C.; Lifshits, E.; Luo, J.; Frangioni, J.V.; Cantley, L.C. Class 1A PI3K regulates vessel integrity during development and tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 9739–9744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo, M.M.; Zelenay, S.; Nye, E.; Castellano, E.; Lassailly, F.; Stamp, G.; Downward, J. RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J. Clin. Investig. 2014, 124, 3601–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, A.; Serra, H.; Pearce, W.; Angulo, A.; Guillermet-Guibert, J.; Friedman, L.S.; Vinals, F.; Gerhardt, H.; Casanovas, O.; Graupera, M.; et al. Inhibition of the p110alpha isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J. Exp. Med. 2013, 210, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, V.; Toh, H.C.; Abastado, J.P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Houghton, A.M.; Mariani, T.J.; Perera, S.; Kim, C.B.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.A.; Hezel, A.; et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Deng, S.; Clowers, M.J.; Velasco, W.V.; Ramos-Castaneda, M.; Moghaddam, S.J. Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 2019, 9, 1556. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [Green Version]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes. Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Mummidi, S.; Cortez, D.M.; Prabhu, S.D.; Valente, A.J.; Chandrasekar, B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J. Physiol. Heart Circ. Physiol. 2008, 294, H2078–H2087. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Baker, J.R.; Vuppusetty, C.; Fenwick, P.; Donnelly, L.E.; Ito, K.; Barnes, P.J. Decreased phosphatase PTEN amplifies PI3K signaling and enhances proinflammatory cytokine release in COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L230–L239. [Google Scholar] [CrossRef] [Green Version]
- Hutti, J.E.; Pfefferle, A.D.; Russell, S.C.; Sircar, M.; Perou, C.M.; Baldwin, A.S. Oncogenic PI3K mutations lead to NF-kappaB-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012, 72, 3260–3269. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110delta prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar]
- Weijzen, S.; Velders, M.P.; Kast, W.M. Modulation of the immune response and tumor growth by activated Ras. Leukemia 1999, 13, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, S.; Sasaki, M.; Scharer, C.D.; Kissick, H.T.; Patterson, D.G.; Magliocca, K.R.; Seykora, J.T.; Sapkota, B.; Gutman, D.A.; Cooper, L.A.; et al. Phosphoinositide 3-Kinase Signaling Can Modulate MHC Class I and II Expression. Mol. Cancer Res. 2019, 17, 2395–2409. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNgamma receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- El-Jawhari, J.J.; El-Sherbiny, Y.M.; Scott, G.B.; Morgan, R.S.; Prestwich, R.; Bowles, P.A.; Blair, G.E.; Tanaka, T.; Rabbitts, T.H.; Meade, J.L.; et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol. Immunol. 2014, 58, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, S.; Wollscheid, U.; Huber, C.; Seliger, B. Multiple levels of MHC class I down-regulation by ras oncogenes. Scand. J. Immunol. 1996, 43, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Bacchetta, R.; Schulz, U.; Roncarolo, M.G. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Granville, C.A.; Memmott, R.M.; Balogh, A.; Mariotti, J.; Kawabata, S.; Han, W.; Lopiccolo, J.; Foley, J.; Liewehr, D.J.; Steinberg, S.M.; et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS ONE 2009, 4, e5061. [Google Scholar] [CrossRef] [PubMed]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant K-Ras Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Bilate, A.M.; Lafaille, J.J. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 2012, 30, 733–758. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.D.; Bork, J.P.; Krueger, B.; Patsenker, E.; Schulze-Krebs, A.; Hahn, E.G.; Schuppan, D. VEGF induces proliferation, migration, and TGF-beta1 expression in mouse glomerular endothelial cells via mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 2005, 334, 1049–1060. [Google Scholar] [CrossRef]
- Lee, S.J.; Yang, E.K.; Kim, S.G. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: Role for Zf9 dephosphorylation. Mol. Pharm. 2006, 70, 415–425. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, S.J.; Kim, S.R.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Lee, Y.R.; Kim, J.S.; Hong, S.J.; et al. Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur. Respir. J. 2008, 31, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018, 103, 1065–1076. [Google Scholar] [CrossRef]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother Cancer 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Sarabi, M.; Pereira Abrantes, M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Jia, L.P.; Liu, Y.; Han, Y.; Deng, Q. Alteration of tumor associated neutrophils by PIK3CA expression in endometrial carcinoma from TCGA data. J. Ovarian. Res. 2019, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef] [Green Version]
- Torphy, R.J.; Zhu, Y.; Schulick, R.D. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann. Gastroenterol. Surg. 2018, 2, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Scilla, K.; Caglevic, C.; Miller, K.; Oliveira, J.; Rolfo, C. Immunotherapy in Lung Cancer: A New Age in Cancer Treatment. Adv. Exp. Med. Biol. 2018, 995, 65–95. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.; de Carne Trecesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wei, F.; Wang, J.; Yu, W.; Shen, M.; Liu, T.; Zhang, D.; Wang, Y.; Ren, X.; Sun, Q. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1(-)PD-L1(+) Bregs through PD-L1/PI3K/AKT/NF-kappaB axis in breast cancer. Cell Death Dis. 2021, 12, 465. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, T.S.; Chan, L.K.; Wong, E.C.; Hui, C.W.; Sneddon, K.; Cheung, T.H.; Yim, S.F.; Lee, J.H.; Yeung, C.S.; Chung, T.K.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFalpha-TGFalpha-EGFR. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.E.; Kandalam, V.; Chakrabarti, S.; Wang, X.; Penninger, J.M.; Davidge, S.T.; Oudit, G.Y.; Kassiri, Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am. J. Physiol. Cell Physiol. 2010, 298, C679–C692. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Bio. Sci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes. Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic K-Ras Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; Pasca di Magliano, M.; Fernandez-Zapico, M.E. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating K-Ras oncogene-induced transformation. J. Biol. Chem. 2013, 288, 11786–11794. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Saotome, T.; Usui, T.; Ohama, T.; Sato, K. Regulation of intestinal myofibroblasts by KRas-mutated colorectal cancer cells through heparin-binding epidermal growth factor-like growth factor. Oncol. Rep. 2017, 37, 3128–3136. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erler, J.T.; Weaver, V.M. Three-dimensional context regulation of metastasis. Clin. Exp. Metastasis 2009, 26, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Cox, T.R.; Bird, D.; Lang, G.; Murray, G.I.; Sun, X.F.; Southall, S.M.; Wilson, J.R.; Erler, J.T. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J. Natl. Cancer Inst. 2011, 103, 407–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Erler, J.T. Lysyl oxidase in colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G659–G666. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Takano, O.; Kato, I.; Takahashi, Y.; Shima, F.; Kataoka, T. Ras inhibitors display an anti-metastatic effect by downregulation of lysyl oxidase through inhibition of the Ras-PI3K-Akt-HIF-1alpha pathway. Cancer Lett. 2017, 410, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Wolfman, J.C.; Wolfman, A. K-ras regulates the steady-state expression of matrix metalloproteinase 2 in fibroblasts. J. Biol. Chem. 2003, 278, 31871–31878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Wang, Q.; Fu, X.H.; Huang, X.H.; Chen, X.L.; Cao, L.Q.; Chen, L.Z.; Tan, H.X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 2009, 39, 177–186. [Google Scholar] [CrossRef]
- Chen, X.C.; Wei, X.T.; Guan, J.H.; Shu, H.; Chen, D. EGF stimulates glioblastoma metastasis by induction of matrix metalloproteinase-9 in an EGFR-dependent mechanism. Oncotarget 2017, 8, 65969–65982. [Google Scholar] [CrossRef] [Green Version]
- Ding, P.; Zhang, X.; Jin, S.; Duan, B.; Chu, P.; Zhang, Y.; Chen, Z.N.; Xia, B.; Song, F. CD147 functions as the signaling receptor for extracellular divalent copper in hepatocellular carcinoma cells. Oncotarget 2017, 8, 51151–51163. [Google Scholar] [CrossRef] [Green Version]
- Kuang, W.; Deng, Q.; Deng, C.; Li, W.; Shu, S.; Zhou, M. Hepatocyte growth factor induces breast cancer cell invasion via the PI3K/Akt and p38 MAPK signaling pathways to up-regulate the expression of COX2. Am. J. Transl. Res. 2017, 9, 3816–3826. [Google Scholar]
- Zhou, R.; Xu, L.; Ye, M.; Liao, M.; Du, H.; Chen, H. Formononetin inhibits migration and invasion of MDA-MB-231 and 4T1 breast cancer cells by suppressing MMP-2 and MMP-9 through PI3K/AKT signaling pathways. Horm Metab. Res. 2014, 46, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Robert, L. Tumors perturbing extracellular matrix biosynthesis. The case of von Recklinghausen’s disease. Pathol. Biol. 2014, 62, 118–122. [Google Scholar] [CrossRef]
- Mehner, C.; Miller, E.; Khauv, D.; Nassar, A.; Oberg, A.L.; Bamlet, W.R.; Zhang, L.; Waldmann, J.; Radisky, E.S.; Crawford, H.C.; et al. Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol. Cancer Res. 2014, 12, 1430–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, K.; Choi, S.; Wyse, M.; Strnadel, J.; Wright, T.; Klemke, R. Eukaryotic Translation Initiation Factor 5A (EIF5A) Regulates Pancreatic Cancer Metastasis by Modulating RhoA and Rho-associated Kinase (ROCK) Protein Expression Levels. J. Biol. Chem. 2015, 290, 29907–29919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta. Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first-in-class KRASinhibitor. Nat. Rev. Drug. Discov. 2021, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef] [PubMed]
- Stalnecker, C.A.; Der, C.J. RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci. Signal 2020, 13, 624. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Krause, G.; Hassenruck, F.; Hallek, M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug. Des. Devel. Ther. 2018, 12, 2577–2590. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Samaniego, F.; Jurczak, W.; Ghosh, N.; Derenzini, E.; Reeves, J.A.; Knopińska-Posłuszny, W.; Cheah, C.Y.; Phillips, T.; Lech-Maranda, E.; et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Saura, C.; Richards, D.A.; Krop, I.E.; Cervantes, A.; Bedard, P.L.; Patel, M.R.; Pusztai, L.; Oliveira, M.; Cardenas, A.K.; et al. Phase II Study of Taselisib (GDC-0032) in Combination with Fulvestrant in Patients with HER2-Negative, Hormone Receptor-Positive Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 4380–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Sanabria Bohorquez, S.M.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.J.; Furman, M.; Winer, E.S. CAL-101: A phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Exp. Opin. Investig. Drug. 2012, 21, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Qayum, N.; Im, J.; Stratford, M.R.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin. Cancer Res. 2012, 18, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.K.; Zhabyeyev, P.; Vanhaesebroeck, B.; Eitzen, G.; Oudit, G.Y.; Moore, R.B.; Murray, A.G. Inactivation of endothelial cell phosphoinositide 3-kinase beta inhibits tumor angiogenesis and tumor growth. Oncogene 2020, 39, 6480–6492. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release 2020, 321, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, C.; Arévalo-Alameda, C.; Castellano, E. The Importance of Being PI3K in the RAS Signaling Network. Genes 2021, 12, 1094. https://doi.org/10.3390/genes12071094
Cuesta C, Arévalo-Alameda C, Castellano E. The Importance of Being PI3K in the RAS Signaling Network. Genes. 2021; 12(7):1094. https://doi.org/10.3390/genes12071094
Chicago/Turabian StyleCuesta, Cristina, Cristina Arévalo-Alameda, and Esther Castellano. 2021. "The Importance of Being PI3K in the RAS Signaling Network" Genes 12, no. 7: 1094. https://doi.org/10.3390/genes12071094
APA StyleCuesta, C., Arévalo-Alameda, C., & Castellano, E. (2021). The Importance of Being PI3K in the RAS Signaling Network. Genes, 12(7), 1094. https://doi.org/10.3390/genes12071094