
The Role of Preclinical Models in Creatine Transporter Deficiency: Neurobiological Mechanisms, Biomarkers and Therapeutic Development

 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Creatine Transporter Deficiency: Clinical Aspects and Therapeutic Management
2.1. Inheritance Pattern and Clinical Signs of CTD
2.2. Diagnostic Methods and Prevalence
2.3. Therapeutic Perspectives in CTD Patients
3. Modeling CTD Features in Animals
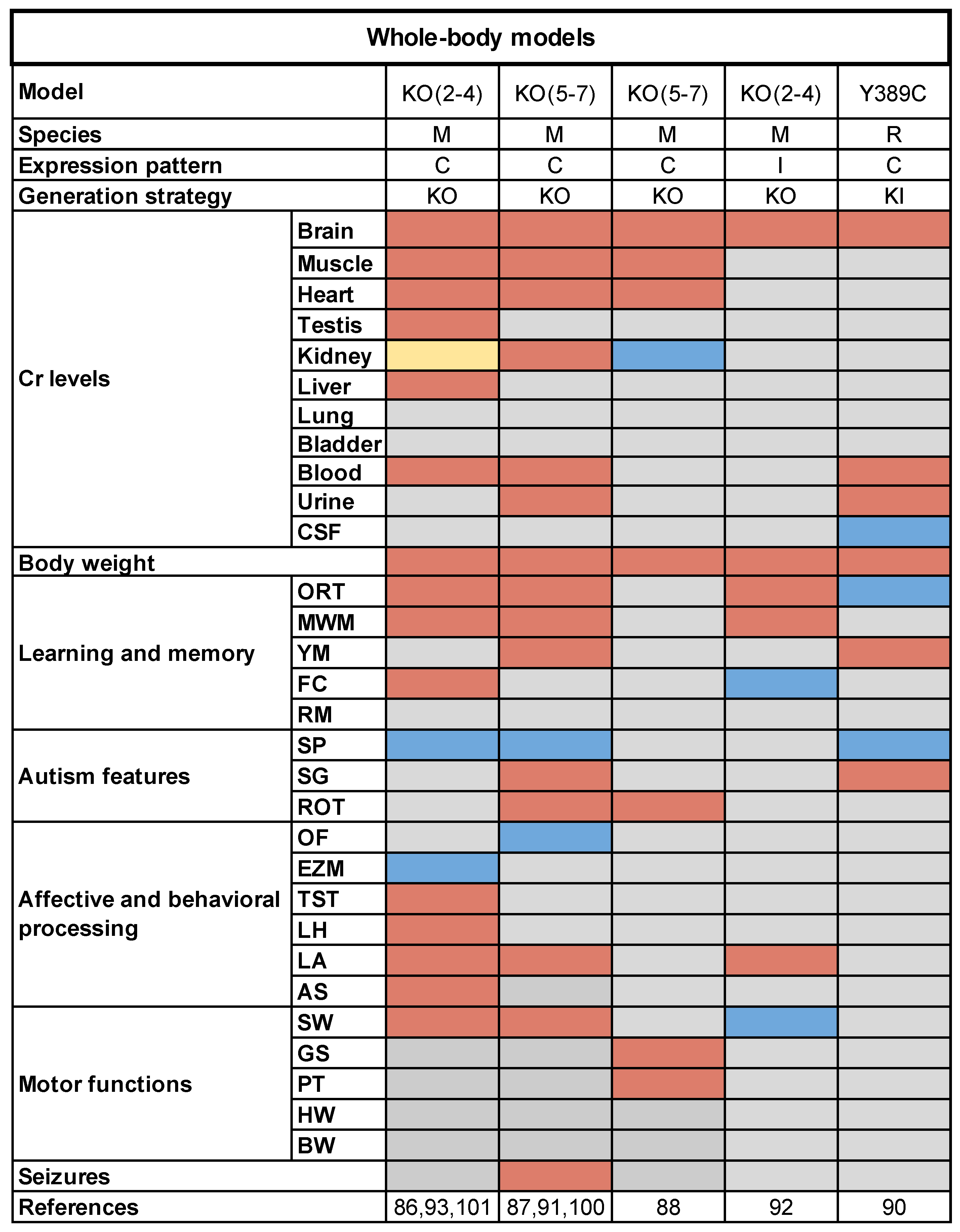
3.1. Whole-Body Rodent Models
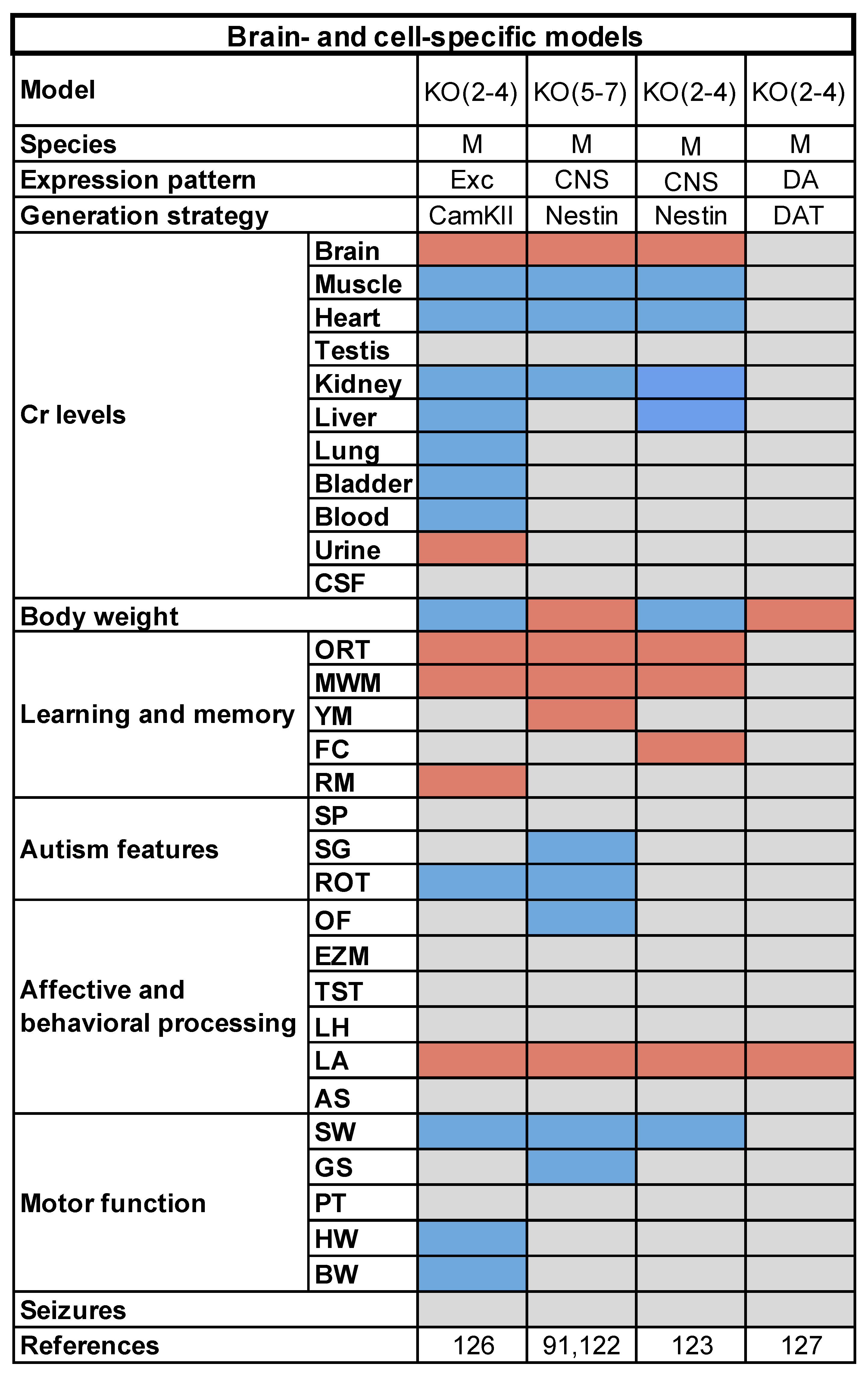
3.2. Brain- and Cell-Specific Conditional Mouse Models
3.3. Preclinical Testing of Potential Therapeutic Strategies
4. Role of Metabolism in Cancer Progression: Novel Insights from Creatine Models
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The Creatine Kinase System and Pleiotropic Effects of Creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [Green Version]
- Ellington, W.R. Evolution and Physiological Roles of Phosphagen Systems. Annu. Rev. Physiol. 2001, 63, 289–325. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.; Kaambre, T.; Guzun, R.; Anmann, T.; Sikk, P.; Schlattner, U.; Wallimann, T.; Aliev, M.; Vendelin, M. The Creatine Kinase Phosphotransfer Network: Thermodynamic and Kinetic Considerations, the Impact of the Mitochondrial Outer Membrane and Modelling Approaches. Subcell. Biochem. 2007, 46, 27–65. [Google Scholar] [PubMed]
- Almeida, L.S.; Salomons, G.S.; Hogenboom, F.; Jakobs, C.; Schoffelmeer, A.N.M. Exocytotic Release of Creatine in Rat Brain. Synapse 2006, 60, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Peral, M.J.; Vázquez-Carretero, M.D.; Ilundain, A.A. Na(+)/Cl(-)/creatine Transporter Activity and Expression in Rat Brain Synaptosomes. Neuroscience 2010, 165, 53–60. [Google Scholar] [CrossRef]
- Neu, A.; Neuhoff, H.; Trube, G.; Fehr, S.; Ullrich, K.; Roeper, J.; Isbrandt, D. Activation of GABA(A) Receptors by Guanidinoacetate: A Novel Pathophysiological Mechanism. Neurobiol. Dis. 2002, 11, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Deyn, P.P.; Marescau, B.; Macdonald, R.L. Guanidino Compounds That Are Increased in Hyperargininemia Inhibit GABA and Glycine Responses on Mouse Neurons in Cell Culture. Epilepsy Res. 1991, 8, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Koga, Y.; Takahashi, H.; Oikawa, D.; Tachibana, T.; Denbow, D.M.; Furuse, M. Brain Creatine Functions to Attenuate Acute Stress Responses through GABAnergic System in Chicks. Neuroscience 2005, 132, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Royes, L.F.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S.; Fiorenza, N.G.; Ferreira, J.; da Silva, A.C.; Priel, M.R.; Ueda, E.S.; Calixto, J.B.; et al. Neuromodulatory Effect of Creatine on Extracellular Action Potentials in Rat Hippocampus: Role of NMDA Receptors. Neurochem. Int. 2008, 53, 33–37. [Google Scholar] [CrossRef]
- Bothwell, J.H.; Rae, C.; Dixon, R.M.; Styles, P.; Bhakoo, K.K. Hypo-Osmotic Swelling-Activated Release of Organic Osmolytes in Brain Slices: Implications for Brain Oedema in Vivo. J. Neurochem. 2001, 77, 1632–1640. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, R.R.; Bonelli, M.A.; Cavazzoni, A.; Brigotti, M.; Fumarola, C.; Sestili, P.; Mozzoni, P.; De Palma, G.; Mutti, A.; Carnicelli, D.; et al. Creatine as a Compatible Osmolyte in Muscle Cells Exposed to Hypertonic Stress. J. Physiol. 2006, 576, 391–401. [Google Scholar] [CrossRef]
- Sestili, P.; Martinelli, C.; Colombo, E.; Barbieri, E.; Potenza, L.; Sartini, S.; Fimognari, C. Creatine as an Antioxidant. Amino Acids 2011, 40, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.M.; Barnes, W.S.; Wu, G.; Song, W.; Demaree, S. Direct Antioxidant Properties of Creatine. Biochem. Biophys. Res. Commun. 2002, 290, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; Brosnan, M.E. Creatine: Endogenous Metabolite, Dietary, and Therapeutic Supplement. Annu. Rev. Nutr. 2007, 27, 241–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosnan, M.E.; Edison, E.E.; da Silva, R.; Brosnan, J.T. New Insights into Creatine Function and Synthesis. Adv. Enzyme Regul. 2007, 47, 252–260. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- Defalco, A.J.; Davies, R.K. The Synthesis of Creatine by the Brain of the Intact Rat. J. Neurochem. 1961, 7, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Hanna-El-Daher, L.; Braissant, O. Creatine Synthesis and Exchanges between Brain Cells: What Can Be Learned from Human Creatine Deficiencies and Various Experimental Models? Amino Acids 2016, 48, 1877–1895. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Henry, H.; Loup, M.; Eilers, B.; Bachmann, C. Endogenous Synthesis and Transport of Creatine in the Rat Brain: An in Situ Hybridization Study. Mol. Brain Res. 2001, 86, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Joncquel-Chevalier Curt, M.; Voicu, P.-M.; Fontaine, M.; Dessein, A.-F.; Porchet, N.; Mention-Mulliez, K.; Dobbelaere, D.; Soto-Ares, G.; Cheillan, D.; Vamecq, J. Creatine Biosynthesis and Transport in Health and Disease. Biochimie 2015, 119, 146–165. [Google Scholar] [CrossRef]
- Nash, S.R.; Giros, B.; Kingsmore, S.F.; Rochelle, J.M.; Suter, S.T.; Gregor, P.; Seldin, M.F.; Caron, M.G. Cloning, Pharmacological Characterization, and Genomic Localization of the Human Creatine Transporter. Receptors Channels 1994, 2, 165–174. [Google Scholar]
- Iyer, G.S.; Krahe, R.; Goodwin, L.A.; Doggett, N.A.; Siciliano, M.J.; Funanage, V.L.; Proujansky, R. Identification of a Testis-Expressed Creatine Transporter Gene at 16p11.2 and Confirmation of the X-Linked Locus to Xq28. Genomics 1996, 34, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Guimbal, C.; Kilimann, M.W. A Na(+)-Dependent Creatine Transporter in Rabbit Brain, Muscle, Heart, and Kidney. cDNA Cloning and Functional Expression. J. Biol. Chem. 1993, 268, 8418–8421. [Google Scholar] [CrossRef]
- Chen, N.-H.; Reith, M.E.A.; Quick, M.W. Synaptic Uptake and beyond: The Sodium- and Chloride-Dependent Neurotransmitter Transporter Family SLC6. Pflugers Arch. 2004, 447, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Béard, E.; Braissant, O. Synthesis and transport of creatine in the CNS: Importance for cerebral functions. J. Neurochem. 2010, 115, 297–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, J.R.; Christie, D.L. Substituted Cysteine Accessibility of the Third Transmembrane Domain of the Creatine Transporter: Defining a Transport Pathway. J. Biol. Chem. 2005, 280, 32649–32654. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Thali, R.F.; Smolak, C.; Gong, F.; Alzamora, R.; Wallimann, T.; Scholz, R.; Pastor-Soler, N.M.; Neumann, D.; Hallows, K.R. Regulation of the Creatine Transporter by AMP-Activated Protein Kinase in Kidney Epithelial Cells. Am. J. Physiol. Renal Physiol. 2010, 299, F167–F177. [Google Scholar] [CrossRef] [Green Version]
- Mak, C.S.W.; Waldvogel, H.J.; Dodd, J.R.; Gilbert, R.T.; Lowe, M.T.J.; Birch, N.P.; Faull, R.L.M.; Christie, D.L. Immunohistochemical Localisation of the Creatine Transporter in the Rat Brain. Neuroscience 2009, 163, 571–585. [Google Scholar] [CrossRef]
- Lowe, M.T.J.; Faull, R.L.M.; Christie, D.L.; Waldvogel, H.J. Distribution of the Creatine Transporter throughout the Human Brain Reveals a Spectrum of Creatine Transporter Immunoreactivity. J. Comp. Neurol. 2015, 523, 699–725. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [Green Version]
- Tasic, B.; Yao, Z.; Graybuck, L.T.; Smith, K.A.; Nguyen, T.N.; Bertagnolli, D.; Goldy, J.; Garren, E.; Economo, M.N.; Viswanathan, S.; et al. Shared and Distinct Transcriptomic Cell Types across Neocortical Areas. Nature 2018, 563, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Hosoya, K.-I. Transport Characteristics of Guanidino Compounds at the Blood-Brain Barrier and Blood-Cerebrospinal Fluid Barrier: Relevance to Neural Disorders. Fluids Barriers CNS 2011, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perasso, L.; Cupello, A.; Lunardi, G.L.; Principato, C.; Gandolfo, C.; Balestrino, M. Kinetics of Creatine in Blood and Brain after Intraperitoneal Injection in the Rat. Brain Res. 2003, 974, 37–42. [Google Scholar] [CrossRef]
- Braissant, O. Creatine and Guanidinoacetate Transport at Blood-Brain and Blood-Cerebrospinal Fluid Barriers. J. Inherit. Metab. Dis. 2012, 35, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fons, C.; Campistol, J. Creatine Defects and Central Nervous System. Semin. Pediatr. Neurol. 2016, 23, 285–289. [Google Scholar] [CrossRef]
- Schulze, A. Creatine Deficiency Syndromes. Mol. Cell. Biochem. 2003, 244, 143–150. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, J.M.; Mancini, G.M.; Salomons, G.S. X-Linked Creatine Transporter Deficiency: Clinical Aspects and Pathophysiology. J. Inherit. Metab. Dis. 2014, 37, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Item, C.B.; Stöckler-Ipsiroglu, S.; Stromberger, C.; Mühl, A.; Alessandrì, M.G.; Bianchi, M.C.; Tosetti, M.; Fornai, F.; Cioni, G. Arginine:glycine Amidinotransferase Deficiency: The Third Inborn Error of Creatine Metabolism in Humans. Am. J. Hum. Genet. 2001, 69, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.C.; Tosetti, M.; Fornai, F.; Alessandri’, M.G.; Cipriani, P.; De Vito, G.; Canapicchi, R. Reversible Brain Creatine Deficiency in Two Sisters with Normal Blood Creatine Level. Ann. Neurol. 2000, 47, 511–513. [Google Scholar] [CrossRef]
- Stöckler, S.; Holzbach, U.; Hanefeld, F.; Marquardt, I.; Helms, G.; Requart, M.; Hänicke, W.; Frahm, J. Creatine Deficiency in the Brain: A New, Treatable Inborn Error of Metabolism. Pediatric Res. 1994, 36, 409–413. [Google Scholar] [CrossRef]
- Salomons, G.S.; van Dooren, S.J.; Verhoeven, N.M.; Cecil, K.M.; Ball, W.S.; Degrauw, T.J.; Jakobs, C. X-Linked Creatine-Transporter Gene (SLC6A8) Defect: A New Creatine-Deficiency Syndrome. Am. J. Hum. Genet. 2001, 68, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecil, K.M.; Salomons, G.S.; Ball, W.S., Jr.; Wong, B.; Chuck, G.; Verhoeven, N.M.; Jakobs, C.; DeGrauw, T.J. Irreversible Brain Creatine Deficiency with Elevated Serum and Urine Creatine: A Creatine Transporter Defect? Ann. Neurol. 2001, 49, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Stockler, S.; Schutz, P.W.; Salomons, G.S. Cerebral Creatine Deficiency Syndromes: Clinical Aspects, Treatment and Pathophysiology. Subcell. Biochem. 2007, 46, 149–166. [Google Scholar] [PubMed]
- Khaikin, Y.; Sidky, S.; Abdenur, J.; Anastasi, A.; Ballhausen, D.; Buoni, S.; Chan, A.; Cheillan, D.; Dorison, N.; Goldenberg, A.; et al. Treatment Outcome of Twenty-Two Patients with Guanidinoacetate Methyltransferase Deficiency: An International Retrospective Cohort Study. Eur. J. Paediatr. Neurol. 2018, 22, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Alessandrì, M.G.; Strigini, F.; Cioni, G.; Battini, R. Increased Creatine Demand during Pregnancy in Arginine: Glycine Amidino-Transferase Deficiency: A Case Report. BMC Pregnancy Childbirth 2020, 20, 506. [Google Scholar] [CrossRef] [PubMed]
- Battini, R.; Alessandrì, M.G.; Casalini, C.; Casarano, M.; Tosetti, M.; Cioni, G. Fifteen-Year Follow-up of Italian Families Affected by Arginine Glycine Amidinotransferase Deficiency. Orphanet J. Rare Dis. 2017, 12, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battini, R.; Alessandrì, M.G.; Leuzzi, V.; Moro, F.; Tosetti, M.; Bianchi, M.C.; Cioni, G. Arginine:glycine Amidinotransferase (AGAT) Deficiency in a Newborn: Early Treatment Can Prevent Phenotypic Expression of the Disease. J. Pediatr. 2006, 148, 828–830. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, J.M.; Betsalel, O.T.; Mercimek-Mahmutoglu, S.; Abulhoul, L.; Grünewald, S.; Anselm, I.; Azzouz, H.; Bratkovic, D.; de Brouwer, A.; Hamel, B.; et al. Phenotype and Genotype in 101 Males with X-Linked Creatine Transporter Deficiency. J. Med. Genet. 2013, 50, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Mancardi, M.M.; Caruso, U.; Schiaffino, M.C.; Baglietto, M.G.; Rossi, A.; Battaglia, F.M.; Salomons, G.S.; Jakobs, C.; Zara, F.; Veneselli, E.; et al. Severe Epilepsy in X-Linked Creatine Transporter Defect (CRTR-D). Epilepsia 2007, 48, 1211–1213. [Google Scholar] [CrossRef]
- Póo-Argüelles, P.; Arias, A.; Vilaseca, M.A.; Ribes, A.; Artuch, R.; Sans-Fito, A.; Moreno, A.; Jakobs, C.; Salomons, G. X-Linked Creatine Transporter Deficiency in Two Patients with Severe Mental Retardation and Autism. J. Inherit. Metab. Dis. 2006, 29, 220–223. [Google Scholar] [CrossRef]
- Arias, A.; Corbella, M.; Fons, C.; Sempere, A.; García-Villoria, J.; Ormazabal, A.; Poo, P.; Pineda, M.; Vilaseca, M.A.; Campistol, J.; et al. Creatine Transporter Deficiency: Prevalence among Patients with Mental Retardation and Pitfalls in Metabolite Screening. Clin. Biochem. 2007, 40, 1328–1331. [Google Scholar] [CrossRef]
- Comeaux, M.S.; Wang, J.; Wang, G.; Kleppe, S.; Zhang, V.W.; Schmitt, E.S.; Craigen, W.J.; Renaud, D.; Sun, Q.; Wong, L.-J. Biochemical, Molecular, and Clinical Diagnoses of Patients with Cerebral Creatine Deficiency Syndromes. Mol. Genet. Metab. 2013, 109, 260–268. [Google Scholar] [CrossRef]
- Bruun, T.U.J.; Sidky, S.; Bandeira, A.O.; Debray, F.-G.; Ficicioglu, C.; Goldstein, J.; Joost, K.; Koeberl, D.D.; Luísa, D.; Nassogne, M.-C.; et al. Treatment Outcome of Creatine Transporter Deficiency: International Retrospective Cohort Study. Metab. Brain Dis. 2018, 33, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Jangid, N.; Surana, P.; Salmonos, G.; Jain, V. Creatine Transporter Deficiency, an Underdiagnosed Cause of Male Intellectual Disability. BMJ Case Rep. 2020, 13. [Google Scholar] [CrossRef]
- Battini, R.; Chilosi, A.; Mei, D.; Casarano, M.; Alessandrì, M.G.; Leuzzi, V.; Ferretti, G.; Tosetti, M.; Bianchi, M.C.; Cioni, G. Mental Retardation and Verbal Dyspraxia in a New Patient with de Novo Creatine Transporter (SLC6A8) Mutation. Am. J. Med. Genet. A 2007, 143A, 1771–1774. [Google Scholar] [CrossRef] [PubMed]
- Battini, R.; Chilosi, A.M.; Casarano, M.; Moro, F.; Comparini, A.; Alessandrì, M.G.; Leuzzi, V.; Tosetti, M.; Cioni, G. Language Disorder with Mild Intellectual Disability in a Child Affected by a Novel Mutation of SLC6A8 Gene. Mol. Genet. Metab. 2011, 102, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Rosenberg, E.H.; Salomons, G.S.; Stroink, H.; van Bokhoven, H.; Hamel, B.C.J.; de Vries, B.B.A. Progressive Intestinal, Neurological and Psychiatric Problems in Two Adult Males with Cerebral Creatine Deficiency Caused by an SLC6A8 Mutation. Clin. Genet. 2005, 68, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.F.; Cecil, K.M. Diagnostic Methods and Recommendations for the Cerebral Creatine Deficiency Syndromes. Pediatr. Res. 2015, 77, 398–405. [Google Scholar] [CrossRef]
- Leuzzi, V.; Mastrangelo, M.; Battini, R.; Cioni, G. Inborn Errors of Creatine Metabolism and Epilepsy. Epilepsia 2013, 54, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Puusepp, H.; Kall, K.; Salomons, G.S.; Talvik, I.; Männamaa, M.; Rein, R.; Jakobs, C.; Õunap, K. The Screening of SLC6A8 Deficiency among Estonian Families with X-Linked Mental Retardation. J. Inherit. Metab. Dis. 2010, 33, 5–11. [Google Scholar] [CrossRef]
- Pyne-Geithman, G.J.; deGrauw, T.J.; Cecil, K.M.; Chuck, G.; Lyons, M.A.; Ishida, Y.; Clark, J.F. Presence of Normal Creatine in the Muscle of a Patient with a Mutation in the Creatine Transporter: A Case Study. Mol. Cell. Biochem. 2004, 262, 35–39. [Google Scholar] [CrossRef] [PubMed]
- deGrauw, T.J.; Cecil, K.M.; Byars, A.W.; Salomons, G.S.; Ball, W.S.; Jakobs, C. The Clinical Syndrome of Creatine Transporter Deficiency. In Guanidino Compounds in Biology and Medicine; Springer: Boston, MA, USA, 2003; pp. 45–48. [Google Scholar]
- Levin, M.D.; Bianconi, S.; Smith, A.; Cawley, N.X.; Do, A.D.; Hammond, D.; Grafstein, J.F.; Thurm, A.; Miller, J.; Perreault, J.; et al. X-Linked Creatine Transporter Deficiency Results in Prolonged QTc and Increased Sudden Death Risk in Humans and Disease Model. Genet. Med. 2021. [Google Scholar] [CrossRef]
- van de Kamp, J.M.; Mancini, G.M.S.; Pouwels, P.J.W.; Betsalel, O.T.; van Dooren, S.J.M.; de Koning, I.; Steenweg, M.E.; Jakobs, C.; van der Knaap, M.S.; Salomons, G.S. Clinical Features and X-Inactivation in Females Heterozygous for Creatine Transporter Defect. Clin. Genet. 2011, 79, 264–272. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Connolly, M.B.; Poskitt, K.J.; Horvath, G.A.; Lowry, N.; Salomons, G.S.; Casey, B.; Sinclair, G.; Davis, C.; Jakobs, C.; et al. Treatment of Intractable Epilepsy in a Female with SLC6A8 Deficiency. Mol. Genet. Metab. 2010, 101, 409–412. [Google Scholar] [CrossRef]
- Cheillan, D.; Joncquel-Chevalier Curt, M.; Briand, G.; Salomons, G.S.; Mention-Mulliez, K.; Dobbelaere, D.; Cuisset, J.-M.; Lion-François, L.; Portes, V.D.; Chabli, A.; et al. Screening for Primary Creatine Deficiencies in French Patients with Unexplained Neurological Symptoms. Orphanet J. Rare Dis. 2012, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrallah, F.; Feki, M.; Kaabachi, N. Creatine and Creatine Deficiency Syndromes: Biochemical and Clinical Aspects. Pediatr. Neurol. 2010, 42, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; van Dooren, S.J.M.; Verhoeven, N.M.; Marsden, D.; Schwartz, C.; Cecil, K.M.; DeGrauw, T.J.; Jakobs, C. X-Linked Creatine Transporter Defect: An Overview. J. Inherit. Metab. Dis. 2003, 26, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Steinbusch, L.K.M.; Wang, P.; Waterval, H.W.A.H.; Stassen, F.A.P.M.; Coene, K.L.M.; Engelke, U.F.H.; Habets, D.D.J.; Bierau, J.; Körver-Keularts, I.M.L.W. Targeted Urine Metabolomics with a Graphical Reporting Tool for Rapid Diagnosis of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.C.; Tosetti, M.; Battini, R.; Leuzzi, V.; Alessandri’, M.G.; Carducci, C.; Antonozzi, I.; Cioni, G. Treatment Monitoring of Brain Creatine Deficiency Syndromes: A 1H- and 31P-MR Spectroscopy Study. AJNR Am. J. Neuroradiol. 2007, 28, 548–554. [Google Scholar] [PubMed]
- Mencarelli, M.A.; Tassini, M.; Pollazzon, M.; Vivi, A.; Calderisi, M.; Falco, M.; Fichera, M.; Monti, L.; Buoni, S.; Mari, F.; et al. Creatine Transporter Defect Diagnosed by Proton NMR Spectroscopy in Males with Intellectual Disability. Am. J. Med. Genet. A 2011, 155A, 2446–2452. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Thomas, R.P.; Bennett, A.; Bianconi, S.; Bruchey, A.; Davis, R.J.; Ficicioglu, C.; Guthrie, W.; Porter, F.D.; Thurm, A. Early Indicators of Creatine Transporter Deficiency. J. Pediatr. 2019, 206, 283–285. [Google Scholar] [CrossRef]
- Clark, A.J.; Rosenberg, E.H.; Almeida, L.S.; Wood, T.C.; Jakobs, C.; Stevenson, R.E.; Schwartz, C.E.; Salomons, G.S. X-Linked Creatine Transporter (SLC6A8) Mutations in about 1% of Males with Mental Retardation of Unknown Etiology. Hum. Genet. 2006, 119, 604–610. [Google Scholar] [CrossRef]
- Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; deGrauw, R.S.; Yntema, H.G.; Bahi, N.; Moraine, C.; Ropers, H.-H.; Fryns, J.-P.; deGrauw, T.J.; et al. High Prevalence of SLC6A8 Deficiency in X-Linked Mental Retardation. Am. J. Hum. Genet. 2004, 75, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DesRoches, C.-L.; Patel, J.; Wang, P.; Minassian, B.; Salomons, G.S.; Marshall, C.R.; Mercimek-Mahmutoglu, S. Estimated Carrier Frequency of Creatine Transporter Deficiency in Females in the General Population Using Functional Characterization of Novel Missense Variants in the SLC6A8 Gene. Gene 2015, 565, 187–191. [Google Scholar] [CrossRef]
- Lion-François, L.; Cheillan, D.; Pitelet, G.; Acquaviva-Bourdain, C.; Bussy, G.; Cotton, F.; Guibaud, L.; Gérard, D.; Rivier, C.; Vianey-Saban, C.; et al. High Frequency of Creatine Deficiency Syndromes in Patients with Unexplained Mental Retardation. Neurology 2006, 67, 1713–1714. [Google Scholar] [CrossRef] [PubMed]
- Yıldız, Y.; Göçmen, R.; Yaramış, A.; Coşkun, T.; Haliloğlu, G. Creatine Transporter Deficiency Presenting as Autism Spectrum Disorder. Pediatrics 2020, 146. [Google Scholar] [CrossRef]
- Stöckler, S.; Hanefeld, F.; Frahm, J. Creatine Replacement Therapy in Guanidinoacetate Methyltransferase Deficiency, a Novel Inborn Error of Metabolism. Lancet 1996, 348, 789–790. [Google Scholar] [CrossRef]
- Schulze, A.; Ebinger, F.; Rating, D.; Mayatepek, E. Improving Treatment of Guanidinoacetate Methyltransferase Deficiency: Reduction of Guanidinoacetic Acid in Body Fluids by Arginine Restriction and Ornithine Supplementation. Mol. Genet. Metab. 2001, 74, 413–419. [Google Scholar] [CrossRef]
- Battini, R.; Leuzzi, V.; Carducci, C.; Tosetti, M.; Bianchi, M.C.; Item, C.B.; Stöckler-Ipsiroglu, S.; Cioni, G. Creatine Depletion in a New Case with AGAT Deficiency: Clinical and Genetic Study in a Large Pedigree. Mol. Genet. Metab. 2002, 77, 326–331. [Google Scholar] [CrossRef]
- Strzelecki, D.; Rabe-Jabłońska, J. Changes in positive and negative symptoms, general psychopathology in schizophrenic patients during augmentation of antipsychotics with glycine: A preliminary 10-week open-label study. Psychiatr. Pol. 2011, 45, 825–837. [Google Scholar]
- Jun, T.; Wennmalm, Å. L-Arginine-Induced Hypotension in the Rat: Evidence That NO Synthesis Is Not Involved. Acta Physiol. Scand. 1994, 152, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Schjelderup, J.; Hope, S.; Vatshelle, C.; van Karnebeek, C.D.M. Treatment Experience in Two Adults with Creatinfe Transporter Deficiency. Mol. Genet. Metab. Rep. 2021, 27, 100731. [Google Scholar] [CrossRef] [PubMed]
- Villar, C.; Campistol, J.; Fons, C.; Armstrong, J.; Mas, A.; Ormazabal, A.; Artuch, R. Glycine and L-Arginine Treatment Causes Hyperhomocysteinemia in Cerebral Creatine Transporter Deficiency Patients. JIMD Rep. 2012, 4, 13–16. [Google Scholar]
- Fons, C.; Sempere, A.; Arias, A.; López-Sala, A.; Póo, P.; Pineda, M.; Mas, A.; Vilaseca, M.A.; Salomons, G.S.; Ribes, A.; et al. Arginine Supplementation in Four Patients with X-Linked Creatine Transporter Defect. J. Inherit. Metab. Dis. 2008, 31, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, M.; Jaggumantri, S.; Sargent, M.; Stockler-Ipsiroglu, S.; van Karnebeek, C.D.M. Treatment of X-Linked Creatine Transporter (SLC6A8) Deficiency: Systematic Review of the Literature and Three New Cases. Mol. Genet. Metab. 2014, 112, 259–274. [Google Scholar] [CrossRef]
- Chilosi, A.; Leuzzi, V.; Battini, R.; Tosetti, M.; Ferretti, G.; Comparini, A.; Casarano, M.; Moretti, E.; Grazia Alessandrì, M.; Cristina Bianchi, M.; et al. Treatment Withl-Arginine Improves Neuropsychological Disorders in a Child with Creatine Transporter Defect. Neurocase 2008, 14, 151–161. [Google Scholar] [CrossRef]
- Chilosi, A.; Casarano, M.; Comparini, A.; Battaglia, F.M.; Mancardi, M.M.; Schiaffino, C.; Tosetti, M.; Leuzzi, V.; Battini, R.; Cioni, G. Neuropsychological Profile and Clinical Effects of Arginine Treatment in Children with Creatine Transport Deficiency. Orphanet J. Rare Dis. 2012, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skelton, M.R.; Schaefer, T.L.; Graham, D.L.; Degrauw, T.J.; Clark, J.F.; Williams, M.T.; Vorhees, C.V. Creatine Transporter (CrT.; Slc6a8) Knockout Mice as a Model of Human CrT Deficiency. PLoS ONE 2011, 6, e16187. [Google Scholar] [CrossRef]
- Baroncelli, L.; Alessandrì, M.G.; Tola, J.; Putignano, E.; Migliore, M.; Amendola, E.; Gross, C.; Leuzzi, V.; Cioni, G.; Pizzorusso, T. A Novel Mouse Model of Creatine Transporter Deficiency. F1000Res. 2014, 3, 228. [Google Scholar] [CrossRef] [Green Version]
- Stockebrand, M.; Sasani, A.; Das, D.; Hornig, S.; Hermans-Borgmeyer, I.; Lake, H.A.; Isbrandt, D.; Lygate, C.A.; Heerschap, A.; Neu, A.; et al. A Mouse Model of Creatine Transporter Deficiency Reveals Impaired Motor Function and Muscle Energy Metabolism. Front. Physiol. 2018, 9, 773. [Google Scholar] [CrossRef]
- Betsalel, O.T.; Pop, A.; Rosenberg, E.H.; Fernandez-Ojeda, M.; Creatine Transporter Research, Group; Jakobs, C.; Salomons, G.S. Detection of Variants in SLC6A8 and Functional Analysis of Unclassified Missense Variants. Mol. Genet. Metab. 2012, 105, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Duran-Trio, L.; Fernandes-Pires, G.; Simicic, D.; Grosse, J.; Roux-Petronelli, C.; Bruce, S.J.; Binz, P.-A.; Sandi, C.; Cudalbu, C.; Braissant, O. A New Rat Model of Creatine Transporter Deficiency Reveals Behavioral Disorder and Altered Brain Metabolism. Sci. Rep. 2021, 11, 1636. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, L.; Molinaro, A.; Cacciante, F.; Alessandrì, M.G.; Napoli, D.; Putignano, E.; Tola, J.; Leuzzi, V.; Cioni, G.; Pizzorusso, T. A Mouse Model for Creatine Transporter Deficiency Reveals Early Onset Cognitive Impairment and Neuropathology Associated with Brain Aging. Hum. Mol. Genet. 2016, 25, 4186–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udobi, K.C.; Delcimmuto, N.; Kokenge, A.N.; Abdulla, Z.I.; Perna, M.K.; Skelton, M.R. Deletion of the Creatine Transporter Gene in Neonatal, but Not Adult, Mice Leads to Cognitive Deficits. J. Inherit. Metab. Dis. 2019, 42, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Wawro, A.M.; Gajera, C.R.; Baker, S.A.; Nirschl, J.J.; Vogel, H.; Montine, T.J. Creatine Transport and Pathological Changes in Creatine Transporter Deficient Mice. J. Inherit. Metab. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bayou, N.; M’rad, R.; Belhaj, A.; Daoud, H.; Zemni, R.; Briault, S.; Helayem, M.B.; Ben Jemaa, L.; Chaabouni, H. The Creatine Transporter Gene Paralogous at 16p11.2 Is Expressed in Human Brain. Comp. Funct. Genomics 2008, 609684. [Google Scholar] [CrossRef]
- Abplanalp, J.; Laczko, E.; Philp, N.J.; Neidhardt, J.; Zuercher, J.; Braun, P.; Schorderet, D.F.; Munier, F.L.; Verrey, F.; Berger, W.; et al. The Cataract and Glucosuria Associated Monocarboxylate Transporter MCT12 Is a New Creatine Transporter. Hum. Mol. Genet. 2013, 22, 3218–3226. [Google Scholar] [CrossRef] [Green Version]
- Loike, J.D.; Zalutsky, D.L.; Kaback, E.; Miranda, A.F.; Silverstein, S.C. Extracellular Creatine Regulates Creatine Transport in Rat and Human Muscle Cells. Proc. Natl. Acad. Sci. USA 1988, 85, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.P.; Ghobrial, L.; Wright, C.R.; Lamon, S.; Brown, E.L.; Kon, M.; Skelton, M.R.; Snow, R.J. Creatine Transporter (SLC6A8) Knockout Mice Display an Increased Capacity for in Vitro Creatine Biosynthesis in Skeletal Muscle. Front. Physiol. 2014, 5, 314. [Google Scholar] [CrossRef] [Green Version]
- Tanila, H. Testing Cognitive Functions in Rodent Disease Models: Present Pitfalls and Future Perspectives. Behav. Brain Res. 2018, 352, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Hautman, E.R.; Kokenge, A.N.; Udobi, K.C.; Williams, M.T.; Vorhees, C.V.; Skelton, M.R. Female Mice Heterozygous for Creatine Transporter Deficiency Show Moderate Cognitive Deficits. J. Inherit. Metab. Dis. 2014, 37, 63–68. [Google Scholar] [CrossRef]
- Mazziotti, R.; Cacciante, F.; Sagona, G.; Lupori, L.; Gennaro, M.; Putignano, E.; Alessandrì, M.G.; Ferrari, A.; Battini, R.; Cioni, G.; et al. Novel Translational Phenotypes and Biomarkers for Creatine Transporter Deficiency. Brain Commun. 2020, 2, fcaa089. [Google Scholar] [CrossRef]
- Abdulla, Z.I.; Pennington, J.L.; Gutierrez, A.; Skelton, M.R. Creatine Transporter Knockout Mice (Slc6a8) Show Increases in Serotonin-Related Proteins and Are Resilient to Learned Helplessness. Behav. Brain Res. 2020, 377, 112254. [Google Scholar] [CrossRef] [PubMed]
- Burt, S.A.; McGue, M.; Krueger, R.F.; Iacono, W.G. Sources of Covariation among the Child-Externalizing Disorders: Informant Effects and the Shared Environment. Psychol. Med. 2005, 35, 1133–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, G.; Rijsdijk, F.; Asherson, P.; Kuntsi, J. Parents and Teachers Make Different Contributions to a Shared Perspective on Hyperactive-Impulsive and Inattentive Symptoms: A Multivariate Analysis of Parent and Teacher Ratings on the Symptom Domains of ADHD. Behav. Genet. 2011, 41, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braissant, O.; Henry, H.; Villard, A.-M.; Speer, O.; Wallimann, T.; Bachmann, C. Creatine Synthesis and Transport during Rat Embryogenesis: Spatiotemporal Expression of AGAT, GAMT and CT1. BMC Dev. Biol. 2005, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braissant, O.; Henry, H.; Béard, E.; Uldry, J. Creatine Deficiency Syndromes and the Importance of Creatine Synthesis in the Brain. Amino Acids 2011, 40, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural Phenotyping Assays for Mouse Models of Autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.E.; Fuccillo, M.V.; Maxeiner, S.; Hayton, S.J.; Gokce, O.; Lim, B.K.; Fowler, S.C.; Malenka, R.C.; Südhof, T.C. Autism-Associated Neuroligin-3 Mutations Commonly Impair Striatal Circuits to Boost Repetitive Behaviors. Cell 2014, 158, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Kalueff, A.V.; Stewart, A.M.; Song, C.; Berridge, K.C.; Graybiel, A.M.; Fentress, J.C. Neurobiology of Rodent Self-Grooming and Its Value for Translational Neuroscience. Nat. Rev. Neurosci. 2016, 17, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, M.S.; Eubanks, J.H. Seizures in Mouse Models of Rare Neurodevelopmental Disorders. Neuroscience 2020, 445, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Fox, S.; Blasi, A.; Elwell, C.E. Illuminating the Developing Brain: The Past, Present and Future of Functional near Infrared Spectroscopy. Neurosci. Biobehav. Rev. 2010, 34, 269–284. [Google Scholar] [CrossRef]
- Bredahl, E.C.; Eckerson, J.M.; Tracy, S.M.; McDonald, T.L.; Drescher, K.M. The Role of Creatine in the Development and Activation of Immune Responses. Nutrients 2021, 13, 751. [Google Scholar] [CrossRef]
- Ji, L.; Zhao, X.; Zhang, B.; Kang, L.; Song, W.; Zhao, B.; Xie, W.; Chen, L.; Hu, X. Slc6a8-Mediated Creatine Uptake and Accumulation Reprogram Macrophage Polarization via Regulating Cytokine Responses. Immunity 2019, 51, 272–284.e7. [Google Scholar] [CrossRef]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.-L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Béard, E.; Torrent, C.; Henry, H. Dissociation of AGAT, GAMT and SLC6A8 in CNS: Relevance to Creatine Deficiency Syndromes. Neurobiol. Dis. 2010, 37, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Hu, X. Microglia/macrophage Polarization: Fantasy or Evidence of Functional Diversity? J. Cereb. Blood Flow Metab. 2020, 40, S134–S136. [Google Scholar] [CrossRef] [PubMed]
- Perna, M.K.; Kokenge, A.N.; Miles, K.N.; Udobi, K.C.; Clark, J.F.; Pyne-Geithman, G.J.; Khuchua, Z.; Skelton, M.R. Creatine Transporter Deficiency Leads to Increased Whole Body and Cellular Metabolism. Amino Acids 2016, 48, 2057–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giusti, L.; Molinaro, A.; Alessandrì, M.G.; Boldrini, C.; Ciregia, F.; Lacerenza, S.; Ronci, M.; Urbani, A.; Cioni, G.; Mazzoni, M.R.; et al. Brain Mitochondrial Proteome Alteration Driven by Creatine Deficiency Suggests Novel Therapeutic Venues for Creatine Deficiency Syndromes. Neuroscience 2019, 409, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Stockebrand, M.; Sauter, K.; Neu, A.; Isbrandt, D.; Choe, C. Differential Regulation of AMPK Activation in Leptin-and Creatine-deficient Mice. FASEB J. 2013, 27, 4147–4156. [Google Scholar] [CrossRef]
- Depino, A.M. Peripheral and Central Inflammation in Autism Spectrum Disorders. Mol. Cell. Neurosci. 2013, 53, 69–76. [Google Scholar] [CrossRef]
- Sherwin, E.; Dinan, T.G.; Cryan, J.F. Recent Developments in Understanding the Role of the Gut Microbiota in Brain Health and Disease. Ann. N. Y. Acad. Sci. 2018, 1420, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Alessandrì, M.G.; Putignano, E.; Leuzzi, V.; Cioni, G.; Baroncelli, L.; Pizzorusso, T. A Nervous System-Specific Model of Creatine Transporter Deficiency Recapitulates the Cognitive Endophenotype of the Disease: A Longitudinal Study. Sci. Rep. 2019, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udobi, K.C.; Kokenge, A.N.; Hautman, E.R.; Ullio, G.; Coene, J.; Williams, M.T.; Vorhees, C.V.; Mabondzo, A.; Skelton, M.R. Cognitive Deficits and Increases in Creatine Precursors in a Brain-Specific Knockout of the Creatine Transporter Gene Slc6a8. Genes Brain Behav. 2018, 17, e12461. [Google Scholar] [CrossRef] [PubMed]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the Glucocorticoid Receptor Gene in the Nervous System Results in Reduced Anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Farr, C.V.; El-Kasaby, A.; Freissmuth, M.; Sucic, S. The Creatine Transporter Unfolded: A Knotty Premise in the Cerebral Creatine Deficiency Syndrome. Front. Synaptic Neurosci. 2020, 12, 588954. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, Y.; Degrauw, T.J.; Lindquist, D.M.; Blanco, V.M.; Pyne-Geithman, G.J.; Daikoku, T.; Chambers, J.B.; Benoit, S.C.; Clark, J.F. Cyclocreatine Treatment Improves Cognition in Mice with Creatine Transporter Deficiency. J. Clin. Invest. 2012, 122, 2837–2846. [Google Scholar] [CrossRef] [Green Version]
- Abdulla, Z.I.; Pahlevani, B.; Lundgren, K.H.; Pennington, J.L.; Udobi, K.C.; Seroogy, K.B.; Skelton, M.R. Deletion of the Creatine Transporter (Slc6a8) in Dopaminergic Neurons Leads to Hyperactivity in Mice. J. Mol. Neurosci. 2020, 70, 102–111. [Google Scholar] [CrossRef]
- Tripp, G.; Wickens, J.R. Neurobiology of ADHD. Neuropharmacology 2009, 57, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Leo, D.; Gainetdinov, R.R. Transgenic Mouse Models for ADHD. Cell Tissue Res. 2013, 354, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, J.N. Translational Animal Models of Autism and Neurodevelopmental Disorders. Dialogues Clin. Neurosci. 2012, 14, 293–305. [Google Scholar] [PubMed]
- Al Dahhan, N.Z.; De Felice, F.G.; Munoz, D.P. Potentials and Pitfalls of Cross-Translational Models of Cognitive Impairment. Front. Behav. Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Enrico, A.; Patrizia, G.; Luisa, P.; Alessandro, P.; Gianluigi, L.; Carlo, G.; Maurizio, B. Electrophysiology and Biochemical Analysis of Cyclocreatine Uptake and Effect in Hippocampal Slices. J. Integr. Neurosci. 2013, 12, 285–297. [Google Scholar] [CrossRef]
- Gorshkov, K.; Wang, A.Q.; Sun, W.; Fisher, E.; Frigeni, M.; Singleton, M.; Thorne, N.; Class, B.; Huang, W.; Longo, N.; et al. Phosphocyclocreatine Is the Dominant Form of Cyclocreatine in Control and Creatine Transporter Deficiency Patient Fibroblasts. Pharm. Res. Perspect. 2019, 7, e00525. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, A.C.; Cohn, M.; Kenyon, G.L. Specificity of Creatine Kinase for Guanidino Substrates. Kinetic and Proton Nuclear Magnetic Relaxation Rate Studies. J. Biol. Chem. 1972, 247, 4382–4388. [Google Scholar] [CrossRef]
- Cacciante, F.; Gennaro, M.; Sagona, G.; Mazziotti, R.; Lupori, L.; Cerri, E.; Putignano, E.; Butt, M.; Do, M.-H.T.; McKew, J.C.; et al. Cyclocreatine Treatment Ameliorates the Cognitive, Autistic and Epileptic Phenotype in a Mouse Model of Creatine Transporter Deficiency. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Annesley, T.M.; Walker, J.B. Cyclocreatine Phosphate as a Substitute for Creatine Phosphate in Vertebrate Tissues. Energistic Considerations. Biochem. Biophys. Res. Commun. 1977, 74, 185–190. [Google Scholar] [CrossRef]
- Mikati, M.A.; Kurdit, R.M.; Rahmeh, A.A.; Farhat, F.; Abu Rialy, S.; Lteif, L.; Francis, E.; Geha, G.; Maraashli, W. Effects of Creatine and Cyclocreatine Supplementation on Kainate Induced Injury in Pre-Pubescent Rats. Brain Inj. 2004, 18, 1229–1241. [Google Scholar] [CrossRef]
- Kale, V.P.; Wallery, J.; Novak, J.; Gibbs, S.; Bourdi, M.; Do, M.-H.T.; McKew, J.C.; Terse, P.S. Evaluation of Chronic Toxicity of Cyclocreatine, a Creatine Analog, in Sprague Dawley Rat after Oral Gavage Administration for up to 26 Weeks. Regul. Toxicol. Pharmacol. 2020, 117, 104750. [Google Scholar] [CrossRef]
- Lunardi, G.; Parodi, A.; Perasso, L.; Pohvozcheva, A.V.; Scarrone, S.; Adriano, E.; Florio, T.; Gandolfo, C.; Cupello, A.; Burov, S.V.; et al. The Creatine Transporter Mediates the Uptake of Creatine by Brain Tissue, but Not the Uptake of Two Creatine-Derived Compounds. Neuroscience 2006, 142, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Du, B. Plasma Esterase Activity and the Metabolism of Drugs with Ester Groups. Ann. N. Y. Acad. Sci. 1971, 179, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Fons, C.; Arias, A.; Sempere, A.; Póo, P.; Pineda, M.; Mas, A.; López-Sala, A.; Garcia-Villoria, J.; Vilaseca, M.A.; Ozaez, L.; et al. Response to Creatine Analogs in Fibroblasts and Patients with Creatine Transporter Deficiency. Mol. Genet. Metab. 2010, 99, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Adriano, E.; Garbati, P.; Damonte, G.; Salis, A.; Armirotti, A.; Balestrino, M. Searching for a Therapy of Creatine Transporter Deficiency: Some Effects of Creatine Ethyl Ester in Brain Slices in Vitro. Neuroscience 2011, 199, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Katseres, N.S.; Reading, D.W.; Shayya, L.; Dicesare, J.C.; Purser, G.H. Non-Enzymatic Hydrolysis of Creatine Ethyl Ester. Biochem. Biophys. Res. Commun. 2009, 386, 363–367. [Google Scholar] [CrossRef]
- Trotier-Faurion, A.; Dézard, S.; Taran, F.; Valayannopoulos, V.; de Lonlay, P.; Mabondzo, A. Synthesis and Biological Evaluation of New Creatine Fatty Esters Revealed Dodecyl Creatine Ester as a Promising Drug Candidate for the Treatment of the Creatine Transporter Deficiency. J. Med. Chem. 2013, 56, 5173–5181. [Google Scholar] [CrossRef]
- Trotier-Faurion, A.; Passirani, C.; Béjaud, J.; Dézard, S.; Valayannopoulos, V.; Taran, F.; de Lonlay, P.; Benoit, J.-P.; Mabondzo, A. Dodecyl Creatine Ester and Lipid Nanocapsule: A Double Strategy for the Treatment of Creatine Transporter Deficiency. Nanomedicine 2015, 10, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Ullio-Gamboa, G.; Udobi, K.C.; Dezard, S.; Perna, M.K.; Miles, K.N.; Costa, N.; Taran, F.; Pruvost, A.; Benoit, J.-P.; Skelton, M.R.; et al. Dodecyl Creatine Ester-Loaded Nanoemulsion as a Promising Therapy for Creatine Transporter Deficiency. Nanomedicine 2019, 14, 1579–1593. [Google Scholar] [CrossRef]
- Adriano, E.; Gulino, M.; Arkel, M.; Salis, A.; Damonte, G.; Liessi, N.; Millo, E.; Garbati, P.; Balestrino, M. Di-Acetyl Creatine Ethyl Ester, a New Creatine Derivative for the Possible Treatment of Creatine Transporter Deficiency. Neurosci. Lett. 2018, 665, 217–223. [Google Scholar] [CrossRef]
- Burov, S.; Leko, M.; Dorosh, M.; Dobrodumov, A.; Veselkina, O. Creatinyl Amino Acids: New Hybrid Compounds with Neuroprotective Activity. J. Pept. Sci. 2011, 17, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Garbati, P.; Adriano, E.; Salis, A.; Ravera, S.; Damonte, G.; Millo, E.; Balestrino, M. Effects of Amide Creatine Derivatives in Brain Hippocampal Slices, and Their Possible Usefulness for Curing Creatine Transporter Deficiency. Neurochem. Res. 2014, 39, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Adriano, E.; Garbati, P.; Salis, A.; Damonte, G.; Millo, E.; Balestrino, M. Creatine Salts Provide Neuroprotection Even after Partial Impairment of the Creatine Transporter. Neuroscience 2017, 340, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kasaby, A.; Kasture, A.; Koban, F.; Hotka, M.; Asjad, H.M.M.; Kubista, H.; Freissmuth, M.; Sucic, S. Rescue by 4-Phenylbutyrate of Several Misfolded Creatine Transporter-1 Variants Linked to the Creatine Transporter Deficiency Syndrome. Neuropharmacology 2019, 161, 107572. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.M.; Berger-Sweeney, J.E.; Eubanks, J.H.; Justice, M.J.; Neul, J.L.; Pozzo-Miller, L.; Blue, M.E.; Christian, D.; Crawley, J.N.; Giustetto, M.; et al. Preclinical Research in Rett Syndrome: Setting the Foundation for Translational Success. Dis. Model. Mech. 2012, 5, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazak, L.; Cohen, P. Creatine Metabolism: Energy Homeostasis, Immunity and Cancer Biology. Nat. Rev. Endocrinol. 2020, 16, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Origins of Cancer: Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.-B. Creatine Kinase in Cell Cycle Regulation and Cancer. Amino Acids 2016, 48, 1775–1784. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK Regulates NADPH Homeostasis to Promote Tumour Cell Survival during Energy Stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, M.; Sun, Y.; Jin, T.; Zhu, P.; Wan, X.; Hou, Y.; Tu, G. SLC6A8-Mediated Intracellular Creatine Accumulation Enhances Hypoxic Breast Cancer Cell Survival via Ameliorating Oxidative Stress. J. Exp. Clin. Cancer Res. 2021, 40, 168. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Guo, X.; Tang, H. SLC6A8 Is Involved in the Progression of Non-Small Cell Lung Cancer through the Notch Signaling Pathway. Ann. Transl. Med. 2021, 9, 264. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, Z.; Yan, H.; Wang, W.; Wu, Z.; Zhang, F.; Zhang, Q.; Shi, G.; Du, J.; Cai, H.; et al. Creatine Promotes Cancer Metastasis through Activation of Smad2/3. Cell Metab. 2021. [Google Scholar] [CrossRef]
- Lillie, J.W.; O’Keefe, M.; Valinski, H.; Hamlin, H.A., Jr.; Varban, M.L.; Kaddurah-Daouk, R. Cyclocreatine (1-Carboxymethyl-2-Iminoimidazolidine) Inhibits Growth of a Broad Spectrum of Cancer Cells Derived from Solid Tumors. Cancer Res. 1993, 53, 3172–3178. [Google Scholar] [PubMed]
- Zarghami, N.; Giai, M.; Yu, H.; Roagna, R.; Ponzone, R.; Katsaros, D.; Sismondi, P.; Diamandis, E.P. Creatine Kinase BB Isoenzyme Levels in Tumour Cytosols and Survival of Breast Cancer Patients. Br. J. Cancer 1996, 73, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramani, M.; Day, B.W.; Schoen, R.E.; Getzenberg, R.H. Altered Expression and Localization of Creatine Kinase B, Heterogeneous Nuclear Ribonucleoprotein F, and High Mobility Group Box 1 Protein in the Nuclear Matrix Associated with Colon Cancer. Cancer Res. 2006, 66, 763–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-H.; Chen, X.-J.; Ou, W.-B.; Zhang, Q.; Lv, Z.-R.; Zhan, Y.; Ma, L.; Huang, T.; Yan, Y.-B.; Zhou, H.-M. Knockdown of Creatine Kinase B Inhibits Ovarian Cancer Progression by Decreasing Glycolysis. Int. J. Biochem. Cell Biol. 2013, 45, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Wu, X.J.; Li, W.C.; Zhuo, C.; Xu, Z.; Tan, C.; Ma, R.; Wang, J.; Pu, J. SLC6A8 Knockdown Suppresses the Invasion and Migration of Human Hepatocellular Carcinoma Huh-7 and Hep3B Cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820983029. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, P.T.; Stracke, M.L.; Nam, S.W.; Woodhouse, E.; O’Keefe, M.; Clair, T.; Liotta, L.A.; Khaddurah-Daouk, R.; Schiffmann, E. Cyclocreatine Inhibits Stimulated Motility in Tumor Cells Possessing Creatine Kinase. Int. J. Cancer 1998, 78, 46–52. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular Metabolic Energetics Can Promote Cancer Progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huddleston, H.G.; Wong, K.-K.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Clinical Applications of Microarray Technology: Creatine Kinase B Is an up-Regulated Gene in Epithelial Ovarian Cancer and Shows Promise as a Serum Marker. Gynecol. Oncol. 2005, 96, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rubery, E.D.; Doran, J.F.; Thompson, R.J. Brain-Type Creatine Kinase BB as a Potential Tumour marker—Serum Levels Measured by Radioimmunoassay in 1015 Patients with Histologically Confirmed Malignancies. Eur. J. Cancer Clin. Oncol. 1982, 18, 951–956. [Google Scholar] [CrossRef]
- Hoosein, N.M.; Martin, K.J.; Abdul, M.; Logothetis, C.J.; Kaddurah-Daouk, R. Antiproliferative Effects of Cyclocreatine on Human Prostatic Carcinoma Cells. Anticancer. Res. 1995, 15, 1339–1342. [Google Scholar] [PubMed]
- Miller, E.E.; Evans, A.E.; Cohn, M. Inhibition of Rate of Tumor Growth by Creatine and Cyclocreatine. Proc. Natl. Acad. Sci. USA 1993, 90, 3304–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Menon, K.; Northey, D.; Liu, J.; Kufe, D.W.; Kaddurah-Daouk, R. Cyclocreatine in Cancer Chemotherapy. Cancer Chemother. Pharmacol. 1995, 35, 411–416. [Google Scholar] [CrossRef]
- Kornacker, M.; Schlattner, U.; Wallimann, T.; Verneris, M.R.; Negrin, R.S.; Kornacker, B.; Staratschek-Jox, A.; Diehl, V.; Wolf, J. Hodgkin Disease-Derived Cell Lines Expressing Ubiquitous Mitochondrial Creatine Kinase Show Growth Inhibition by Cyclocreatine Treatment Independent of Apoptosis. Int. J. Cancer 2001, 94, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnhardt, F.-G.; Bock, C.; Röhn, G.; Ernestus, R.-I.; Hoehn, M. Metabolic Differences between Primary and Recurrent Human Brain Tumors: A 1H NMR Spectroscopic Investigation. NMR Biomed. 2005, 18, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Onda, T.; Uzawa, K.; Endo, Y.; Bukawa, H.; Yokoe, H.; Shibahara, T.; Tanzawa, H. Ubiquitous Mitochondrial Creatine Kinase Downregulated in Oral Squamous Cell Carcinoma. Br. J. Cancer 2006, 94, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Bisbal, M.C.; Celda, B. Proton Magnetic Resonance Spectroscopy Imaging in the Study of Human Brain Cancer. Q. J. Nucl. Med. Mol. Imaging 2009, 53, 618–630. [Google Scholar] [PubMed]
- Patra, S.; Ghosh, A.; Roy, S.S.; Bera, S.; Das, M.; Talukdar, D.; Ray, S.; Wallimann, T.; Ray, M. A Short Review on Creatine–creatine Kinase System in Relation to Cancer and Some Experimental Results on Creatine as Adjuvant in Cancer Therapy. Amino Acids 2012, 42, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Ma, X.; Wang, X.; Yu, J.; Wang, Y.-C.; Smith, D.J.; Zhou, Y.; Li, Z.; Kim, Y.J.; Clarke, N.; et al. Creatine Uptake Regulates CD8 T Cell Antitumor Immunity. J. Exp. Med. 2019, 216, 2869–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghirardini, E.; Calugi, F.; Sagona, G.; Di Vetta, F.; Palma, M.; Battini, R.; Cioni, G.; Pizzorusso, T.; Baroncelli, L. The Role of Preclinical Models in Creatine Transporter Deficiency: Neurobiological Mechanisms, Biomarkers and Therapeutic Development. Genes 2021, 12, 1123. https://doi.org/10.3390/genes12081123
Ghirardini E, Calugi F, Sagona G, Di Vetta F, Palma M, Battini R, Cioni G, Pizzorusso T, Baroncelli L. The Role of Preclinical Models in Creatine Transporter Deficiency: Neurobiological Mechanisms, Biomarkers and Therapeutic Development. Genes. 2021; 12(8):1123. https://doi.org/10.3390/genes12081123
Chicago/Turabian StyleGhirardini, Elsa, Francesco Calugi, Giulia Sagona, Federica Di Vetta, Martina Palma, Roberta Battini, Giovanni Cioni, Tommaso Pizzorusso, and Laura Baroncelli. 2021. "The Role of Preclinical Models in Creatine Transporter Deficiency: Neurobiological Mechanisms, Biomarkers and Therapeutic Development" Genes 12, no. 8: 1123. https://doi.org/10.3390/genes12081123
APA StyleGhirardini, E., Calugi, F., Sagona, G., Di Vetta, F., Palma, M., Battini, R., Cioni, G., Pizzorusso, T., & Baroncelli, L. (2021). The Role of Preclinical Models in Creatine Transporter Deficiency: Neurobiological Mechanisms, Biomarkers and Therapeutic Development. Genes, 12(8), 1123. https://doi.org/10.3390/genes12081123