
Diversity and Paleodemography of the Addax (Addax nasomaculatus), a Saharan Antelope on the Verge of Extinction

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
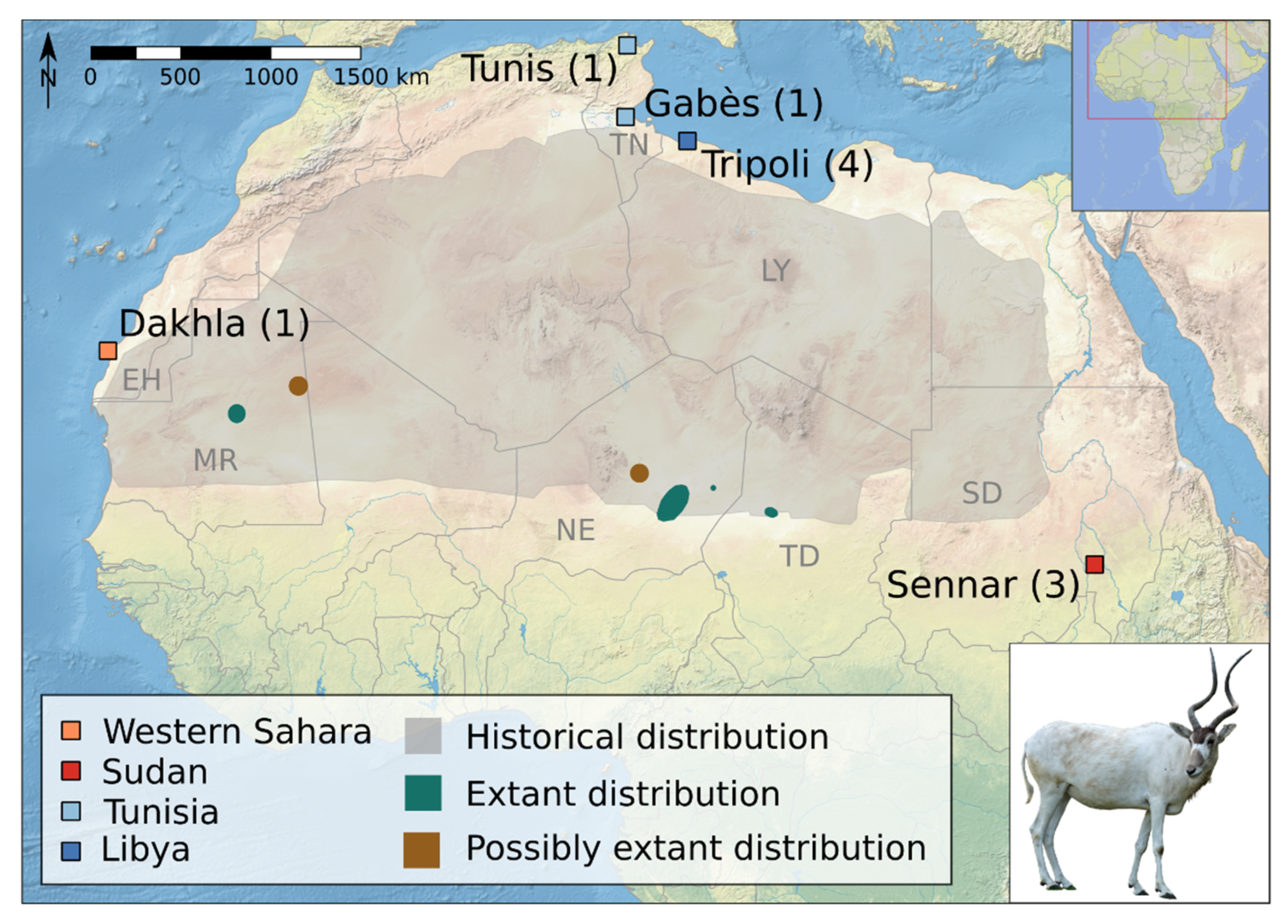
2.1. Samples
2.2. Laboratory Procedures
2.2.1. Nuclear Genome
2.2.2. Mitochondrial Genomes
2.3. Bioinformatic Procedures and Analyses
2.3.1. Nuclear Genome
2.3.2. Mitochondrial Genomes
3. Results
3.1. Assembly of Nuclear Genome and Mitochondrial Genome Data
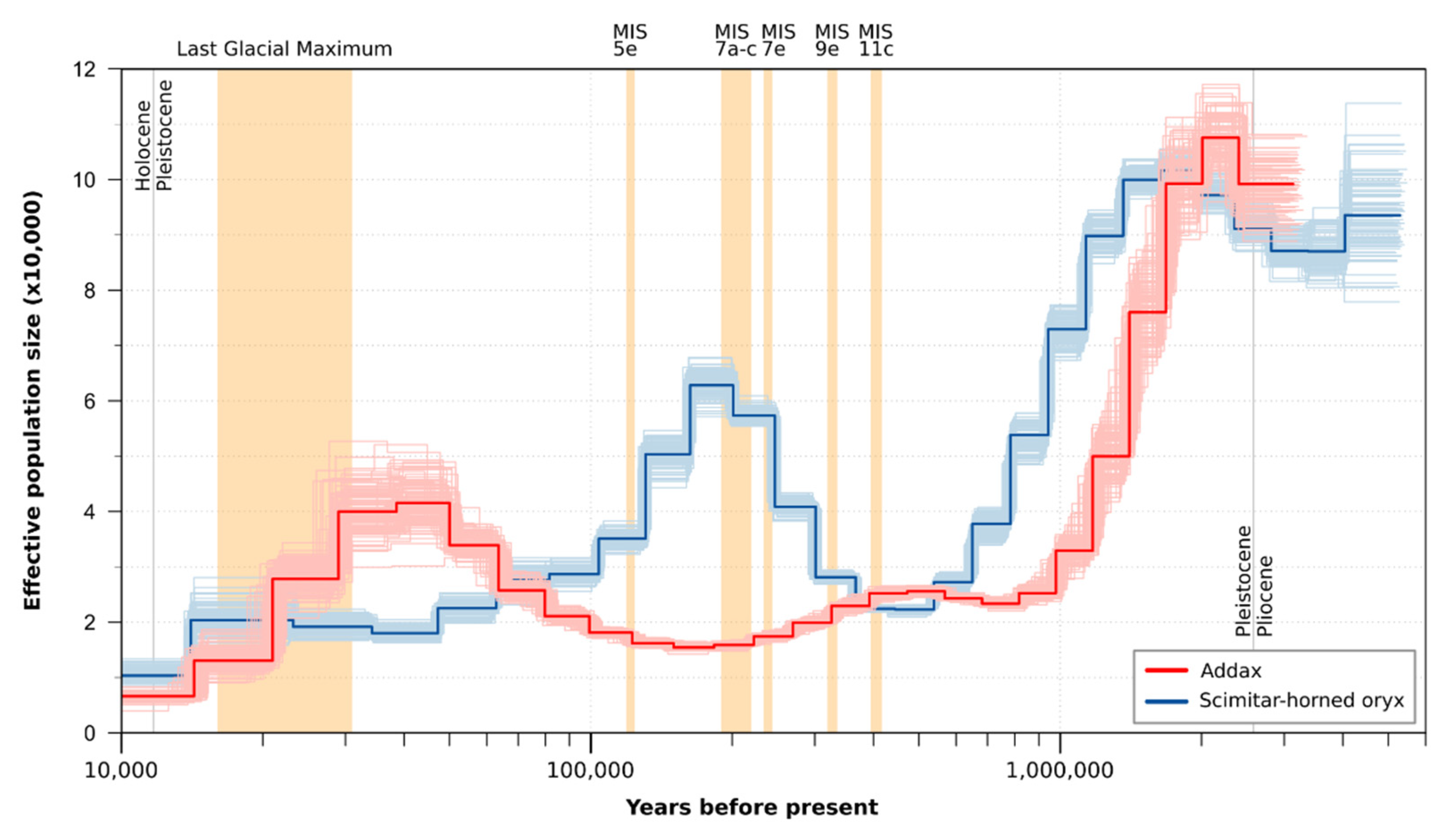
3.1.1. Genetic Diversity and Demographic History
3.1.2. Inbreeding Assessment
3.1.3. Pairwise Sequential Markovian Coalescent Model
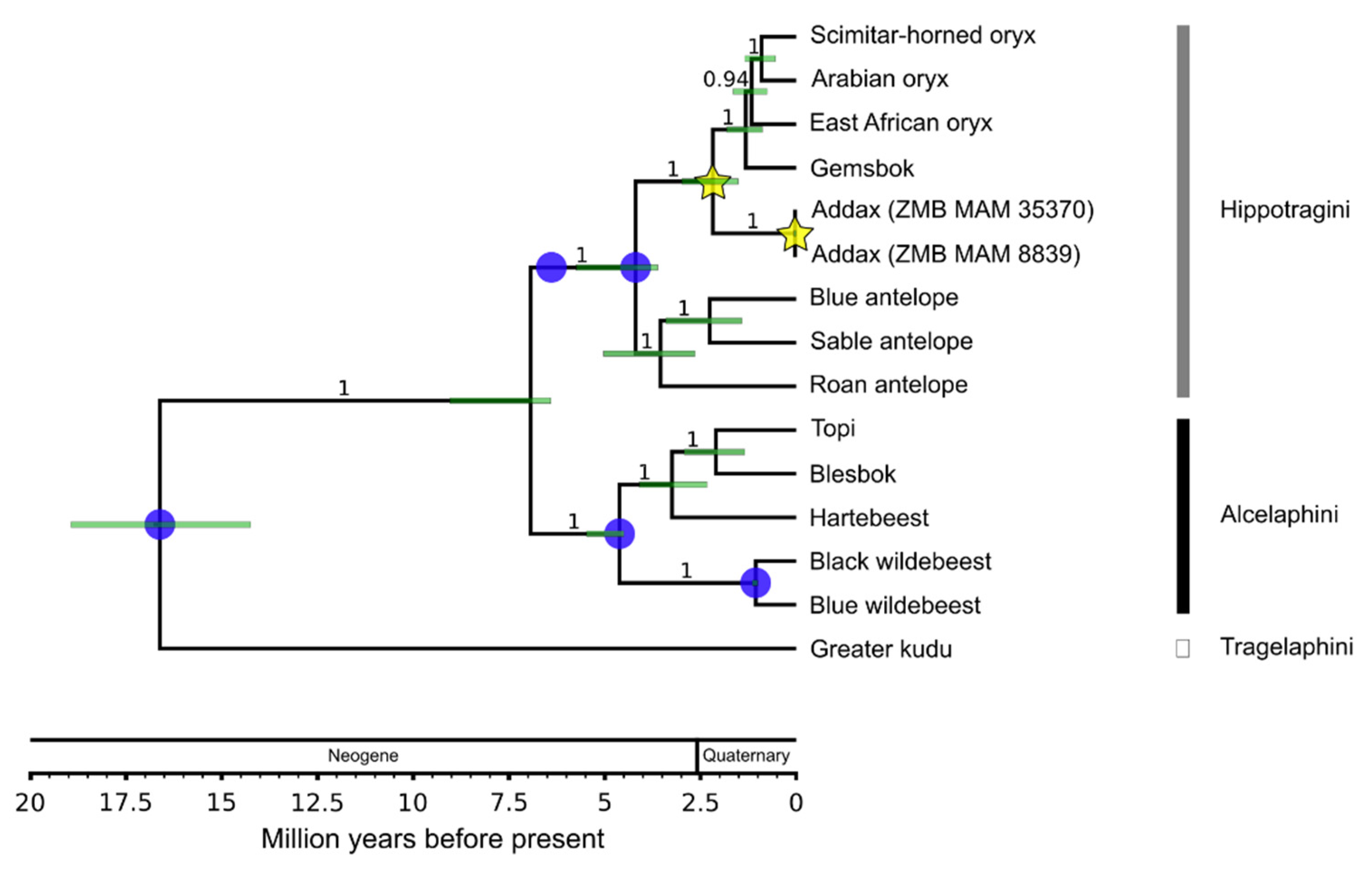
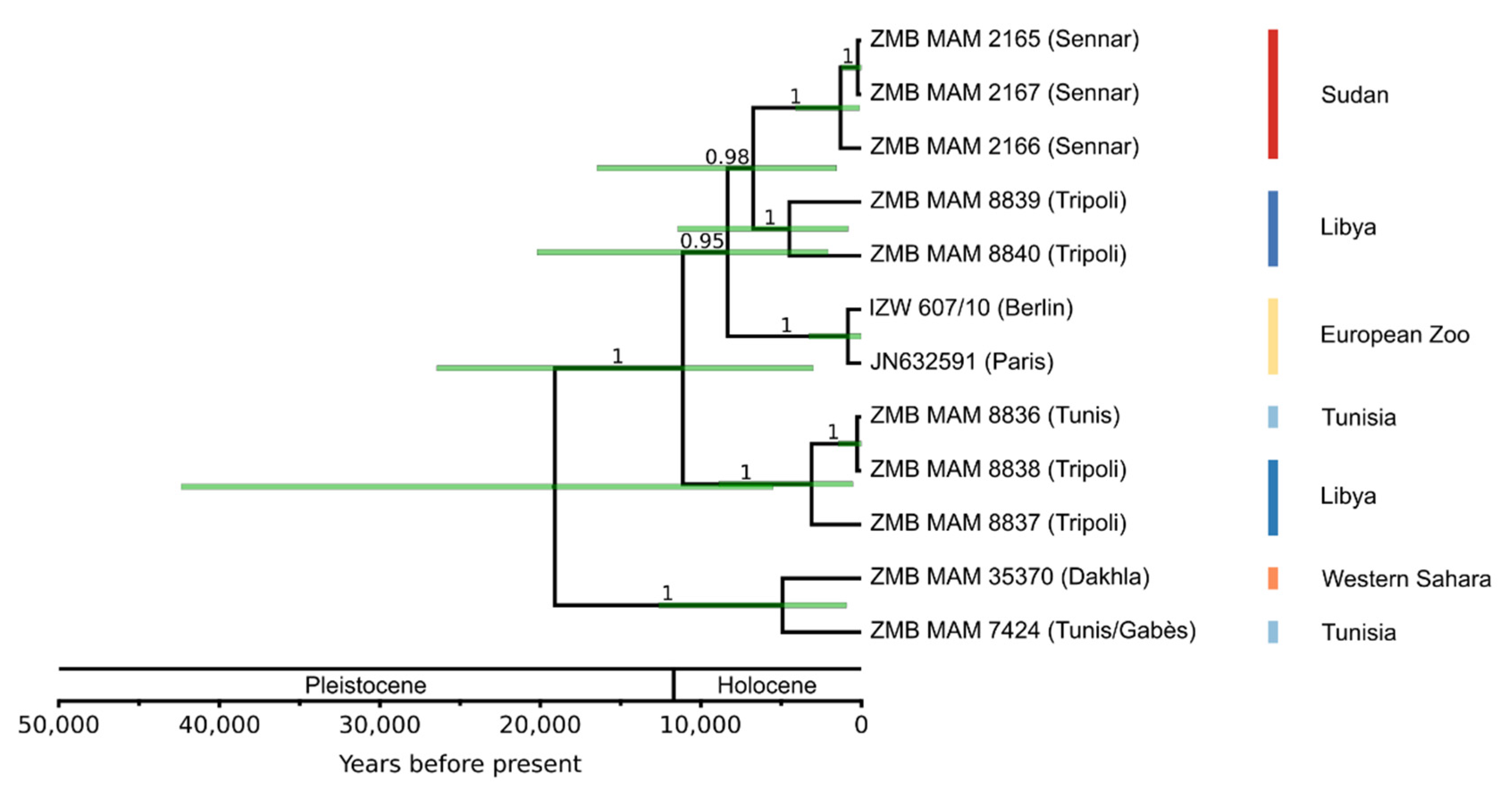
3.2. Divergence Age and Phylogeographic Structure
3.2.1. Species Phylogeny
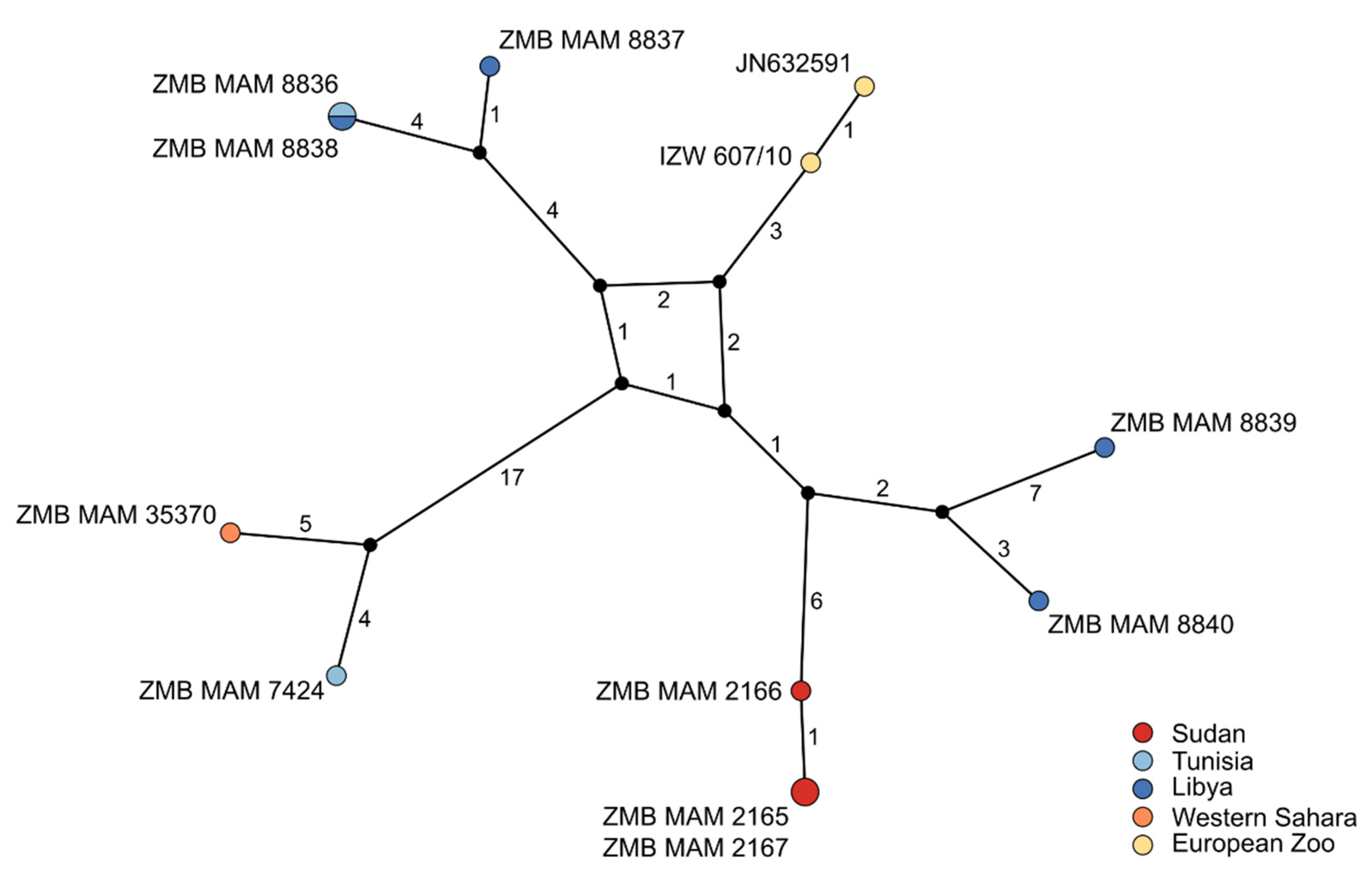
3.2.2. Population Phylogeny
4. Discussion
4.1. The Addax Exibits Low Nuclear and Intermediate Mitochondrial Diversity
4.2. Weak Phylogeographic Structure and Climatic Influence on Population History
4.3. Conservation Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IUCN SSC Antelope Specialist Group. Addax nasomaculatus. IUCN Red List Threat. Species 2016, e.T512A50180603. [Google Scholar] [CrossRef]
- Dolan, J., Jr. Notes on Addax nasomaculatus (De Blainville, 1816). Z. Säugetierkd. 1966, 31, 23–31. [Google Scholar]
- Newby, J. Can addax and oryx be saved in the Sahel? Oryx 1980, 15, 262–266. [Google Scholar] [CrossRef]
- Beudels-Jamar, R.C.; Devillers, P.; Lafontaine, R.-M.; Newby, J. Addax nasomaculatus. In Sahelo-Saharan Antelopes. Status and Perspectives. Report on the Conservation Status of the Six Sahelo-Saharan Antelopes; Beudels, R.C., Devillers, P., Lafontaine, R.-M., Devillers-Terschuren, J., Beudels, M.-O., Eds.; CMS SSA Concerted Action (2nd ed.); CMS Technical Series Publication No. 11; UNEP/CMS Secretariat: Bonn, Germany, 2005; pp. 39–56. [Google Scholar]
- Newby, J. Addax nasomaculatus Addax. In Mammals of Africa; Bloomsbury: London, UK; New Dehli, India; New York, NY, USA; Sydney, NSW, Australia, 2013; Volume 6, pp. 566–571. [Google Scholar]
- Durant, S.M.; Wacher, T.; Bashir, S.; Woodroffe, R.; De Ornellas, P.; Ransom, C.; Newby, J.; Abáigar, T.; Abdelgadir, M.; El Alqamy, H.; et al. Fiddling in biodiversity hotspots while deserts burn? Collapse of the Sahara’s megafauna. Divers. Distrib. 2014, 20, 114–122. [Google Scholar] [CrossRef]
- Rabeil, T. Addax survey in Niger. Sandscript 2016, 19, 6. [Google Scholar]
- DCFAP & DFCPR. Plan d’Action Regional pur l’Addax et la Gazelle Dama 2018–2022; Mallon, D., Pinchon, S., Eds.; NOÉ: Paris, France, 2017. [Google Scholar]
- Chardonnet, P.; Mallon, D.; Woodfine, T. To Save the Addax Antelope, the Oil Sector and Government Must Work Together with Conservationists. Available online: https://www.iucn.org/crossroads-blog/202011/save-addax-antelope-oil-sector-and-government-must-work-together-conservationists (accessed on 7 January 2021).
- Wacher, T.J.; Newby, J.E.; Monfort, S.L.; Tubiana, J.; Moksia, D.; Houston, W.; Dixon, A.M. Sahelo-Saharan interest group antelope update Chad 2001 and Niger 2002. In Antelope Survey Update. Number 9: November 2004. IUCN/SSC Antelope Specialist Group Report; Chardonnet, B., Chardonnet, P., Eds.; IUCN: Gland, Switzerland, 2004; pp. 52–59. [Google Scholar]
- Wildt, D.; Miller, P.; Koepfli, K.-P.; Pukazhenthi, B.; Palfrey, K.; Livingston, G.; Beetem, D.; Shurter, S.; Gregory, J.; Takács, M.; et al. Breeding centers, private ranches, and genomics for creating sustainable wildlife populations. BioScience 2019, 69, 928–943. [Google Scholar] [CrossRef]
- Riordan, P.; Gilbert, T.C.; Petretto, M.; Craig, M.S.; Banfield, L.; Ivy, J.; Senn, H.; Abid, H.; Nouioui, M. The genetic diversity of addax Addax nasomaculatus in reintroduced and ex-situ managed populations. Gnusletter 2017, 34, 21. [Google Scholar]
- Spatz, P. Meine Reise nach Rio de oro. Reisebericht und Beobachtungen an Säugetieren. Z. Säugetierkd. 1926, 1, 23–28. [Google Scholar]
- Newby, J.E. Aridland wildlife in decline: The case of the scimitar-horned oryx. In Conservation and Biology of Desert Antelopes; Dixon, A., Jones, D., Eds.; Christopher Helm: London, UK, 1988; pp. 146–162. [Google Scholar]
- IUCN Saharan Addax Antelope Faces Imminent Extinction. Available online: https://www.iucn.org/content/saharan-addax-antelope-faces-imminent-extinction-0 (accessed on 1 October 2020).
- Brito, J.C.; Durant, S.M.; Pettorelli, N.; Newby, J.; Canney, S.; Algadafi, W.; Rabeil, T.; Crochet, P.-A.; Pleguezuelos, J.M.; Wacher, T.; et al. Armed conflicts and wildlife decline: Challenges and recommendations for effective conservation policy in the Sahara-Sahel. Conserv. Lett. 2018, 11, e12446. [Google Scholar] [CrossRef]
- Gilbert, T.; Woodfine, T.; Petretto, M.; Nouioui, M.; Houston, B.; Riordan, P. Reintroduction of addax to Djebil National Park, Tunisia. In Global Reintroduction Perspectives: 2018. Case Studies from around the Globe; Soorae, P.S., Ed.; IUCN, International Union for Conservation of Nature & Environment Agency: Gland & Abu Dhabi, United Arab Emirates, 2018; pp. 120–124. [Google Scholar]
- Ewi, M. A decade of kidnappings and terrorism in West Africa and the Trans-Sahel region. Afr. Secur. Rev. 2010, 19, 64–71. [Google Scholar] [CrossRef]
- Lohmann, A. Who Owns the Sahara?: Old Conflicts, New Menaces: Mali and the Central Sahara between the Tuareg, Al Qaida and Organized Crime; Friedrich-Ebert-Stiftung: Abuja, Nigeria, 2011. [Google Scholar]
- Sahara Conservation Fund. Niger’s addax hit by drought & oil. Sandscript 2010, 8, 5. [Google Scholar]
- Newby, J. Niger—The Aïr-Ténéré National Nature Reserve. In Living with Wildlife: Wildlife Resource Management with Local Participation in Africa; Kiss, A., Ed.; World Bank Technical Paper; The World Bank: Washington, DC, USA, 1990; pp. 53–58. [Google Scholar]
- Balout, L. Note sur la preśence de restes fossiles d’une Addax nasomaculata Blainv. parmi des ossements découverts au Parc d’Hydra, Commune de Birmandreis (Alger). Bull. Société d’Histoire Nat. L’Afrique Nord 1942, 33, 138–140. [Google Scholar]
- Thomas, H. La faune de la Grotte à Néandertaliens du Jebel Irhoud (Maroc). Quaterenaria 1981, 23, 191–217. [Google Scholar]
- Peters, J. The faunal remains collected by the Bagnold-Mond Expedition in the Gilf Kebir and Jebel Uweinat in 1938. Archéologie Du Nil Moyen 1987, 2, 251–264. [Google Scholar]
- Krausman, P.R.; Casey, A.L. Addax nasomaculatus. Mamm. Species 2007, 807, 1–4. [Google Scholar] [CrossRef]
- Gentry, A.W. Bovidae. In Cenozoic Mammals of Africa; Werdelin, L., Ed.; University of California Press: Berkeley, CA, USA, 2010; pp. 741–796. [Google Scholar]
- Bibi, F. A multi-calibrated mitochondrial phylogeny of extant Bovidae (Artiodactyla, ruminantia) and the importance of the fossil record to systematics. BMC Evol. Biol. 2013, 13, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, J.C.; Godinho, R.; Martínez-Freiría, F.; Pleguezuelos, J.M.; Rebelo, H.; Santos, X.; Vale, C.G.; Velo-Antón, G.; Boratyński, Z.; Carvalho, S.B.; et al. Unravelling biodiversity, evolution and threats to conservation in the Sahara-Sahel. Biol. Rev. 2014, 89, 215–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.; Poulsen, L.; Schulte-Herbrüggen, B.; Mackinnon, K.; Crawhall, N.; Henwood, W.D.; Dudley, N.; Smith, J.; Gudka, M. Conserving Dryland Biodiversity; IUCN: Nairobi, Kenya, 2012. [Google Scholar]
- Durant, S.M.; Pettorelli, N.; Bashir, S.; Woodroffe, R.; Wacher, T.; De Ornellas, P.; Ransom, C.; Abàigar, T.; Abdelgadir, M.; El Alqamy, H.; et al. Forgotten biodiversity in desert ecosystems. Science 2012, 336, 1379–1380. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef]
- Fortes, G.G.; Paijmans, J.L.A. Analysis of whole mitogenomes from ancient samples. In Whole Genome Amplification. Methods in Molecular Biology; Kroneis, T., Ed.; Methods in Molecular Biology; Humana Press: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2015; Volume 1347, pp. 179–195. [Google Scholar]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohland, N.; Siedel, H.; Hofreiter, M. Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens. BioTechniques 2004, 36, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Taron, U.H.; Lell, M.; Barlow, A.; Paijmans, J.L.A. Testing of alignment parameters for ancient samples: Evaluating and optimizing mapping parameters for ancient samples using the TAPAS tool. Genes 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansauge, M.-T.; Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Delsuc, F.; Ropiquet, A.; Hammer, C.; Jansen van Vuuren, B.; Matthee, C.; Ruiz-Garcia, M.; Catzeflis, F.; Areskoug, V.; Nguyen, T.T.; et al. Pattern and timing of diversification of cetartiodactyla (Mammalia, laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. C. R. Biol. 2012, 335, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Rooks, M.; Xuan, Z.; Bhattacharjee, A.; Gordon, D.B.; Brizuela, L.; McCombie, W.R.; Hannon, G.J. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 2009, 4, 960–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. BFC: Correcting Illumina sequencing errors. Bioinformatics 2015, 31, 2885–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.; Du, X.; Zhang, W.; Tosser-Klopp, G.; Wang, J.; Yang, S.; Liang, J.; et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Zhang, X.; Xie, M.; Arefnezhad, B.; Wang, Z.; Wang, W.; Feng, S.; Huang, G.; Guan, R.; Shen, W.; et al. Reference genome of wild goat (Capra aegagrus) and sequencing of goat breeds provide insight into genic basis of goat domestication. BMC Genom. 2015, 16, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, J.H.; Hackl, T.; Koepfli, K.-P.; Hofreiter, M. Improving draft genome contiguity with reference-derived in silico mate-pair libraries. GigaScience 2018, 7, giy029. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremme, G.; Steinbiss, S.; Kurtz, S. GenomeTools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Humble, E.; Dobrynin, P.; Senn, H.; Chuven, J.; Scott, A.F.; Mohr, D.W.; Dudchenko, O.; Omer, A.D.; Colaric, Z.; Aiden, E.L.; et al. Chromosomal-level genome assembly of the scimitar-horned oryx: Insights into diversity and demography of a species extinct in the wild. Mol. Ecol. Resour. 2020, 20, 1668–1681. [Google Scholar] [CrossRef]
- Prasad, A.; Lorenzen, E.D.; Westbury, M.V. Evaluating the role of reference-genome phylogenetic distance on evolutionary inference. Mol. Ecol. Resour. 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Russell, P.; Meyer, M.; Mauceli, E.; Alföldi, J.; Di Palma, F.; Lindblad-Toh, K. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 2010, 26, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Krause, F. EEP Addax Addax nasomaculatus—Population Analysis 2020; Zoo Hannover: Hannover, Germany, 2020. [Google Scholar]
- Gilbert, T.; Langenhorst, T. International Studbook for the Scimitar-Horned Oryx Oryx dammah, 14th ed.; Marwell Wildlife: Winchester, UK, 2019. [Google Scholar]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2021. [Google Scholar]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.; Li, Z.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef]
- Farré, M.; Li, Q.; Zhou, Y.; Damas, J.; Chemnick, L.G.; Kim, J.; Ryder, O.A.; Ma, J.; Zhang, G.; Larkin, D.M.; et al. A near-chromosome-scale genome assembly of the gemsbok (Oryx gazella): An iconic antelope of the Kalahari Desert. GigaScience 2019, 8, giy162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepfli, K.-P.; Tamazian, G.; Wildt, D.; Dobrynin, P.; Kim, C.; Frandsen, P.B.; Godinho, R.; Yurchenko, A.A.; Komissarov, A.; Krasheninnikova, K.; et al. Whole genome sequencing and re-sequencing of the sable antelope (Hippotragus niger): A resource for monitoring diversity in ex situ and in situ populations. G3 2019, 9, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westbury, M.V.; Hartmann, S.; Barlow, A.; Wiesel, I.; Leo, V.; Welch, R.; Parker, D.M.; Sicks, F.; Ludwig, A.; Dalén, L.; et al. Extended and continuous decline in effective population size results in low genomic diversity in the world’s rarest hyena species, the brown hyena. Mol. Biol. Evol. 2018, 35, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Improving SNP discovery by base alignment quality. Bioinformatics 2011, 27, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Westbury, M.V.; Le Duc, D.; Duchêne, D.A.; Krishnan, A.; Prost, S.; Rutschmann, S.; Grau, J.H.; Dalen, L.; Weyrich, A.; Norén, K.; et al. Ecological specialisation and evolutionary reticulation in extant Hyaenidae. Mol. Biol. Evol. 2021, msab055. [Google Scholar] [CrossRef]
- Renaud, G.; Hanghøj, K.; Korneliussen, T.S.; Willerslev, E.; Orlando, L. Joint estimates of heterozygosity and runs of homozygosity for modern and ancient samples. Genetics 2019, 212, 587–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Ropiquet, A.; Couloux, A.; Cruaud, C. Evolution of the mitochondrial genome in mammals living at high altitude: New insights from a study of the tribe Caprini (Bovidae, antilopinae). J. Mol. Evol. 2009, 68, 293–310. [Google Scholar] [CrossRef]
- Steiner, C.C.; Charter, S.J.; Houck, M.L.; Ryder, O.A. Molecular phylogeny and chromosomal evolution of Alcelaphini (Antilopinae). J. Hered. 2014, 105, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Hempel, E.; Bibi, F.; Faith, J.T.; Brink, J.S.; Kalthoff, D.C.; Kamminga, P.; Paijmans, J.L.A.; Westbury, M.V.; Hofreiter, M.; Zachos, F.E. Identifying the true number of specimens of the extinct blue antelope (Hippotragus leucophaeus). Sci. Rep. 2021, 11, 2100. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Snell, Q.; Walke, P.; Posada, D.; Crandall, K. TCS: Estimating gene genealogies. In Proceedings of the 16th International Parallel and Distributed Processing Symposium, Ft. Lauderdale, FL, USA, 15–19 April 2002; IEEE: Piscataway, NJ, USA, 2002. [Google Scholar]
- Douglas, K.C.; Halbert, N.D.; Kolenda, C.; Childers, C.; Hunter, D.L.; Derr, J.N. Complete mitochondrial DNA sequence analysis of Bison bison and bison–cattle hybrids: Function and phylogeny. Mitochondrion 2011, 11, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Achilli, A.; Olivieri, A.; Soares, P.; Lancioni, H.; Kashani, B.H.; Perego, U.A.; Nergadze, S.G.; Carossa, V.; Santagostino, M.; Capomaccio, S.; et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc. Natl. Acad. Sci. USA 2012, 109, 2449–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, R.; Brüniche-Olsen, A.; Siegismund, H.R. Cape buffalo mitogenomics reveals a Holocene shift in the African human-megafauna dynamics. Mol. Ecol. 2012, 21, 3947–3959. [Google Scholar] [CrossRef] [PubMed]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jónsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary genomics and conservation of the endangered Przewalski’s horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef] [Green Version]
- Themudo, G.E.; Rufino, A.C.; Campos, P.F. Complete mitochondrial DNA sequence of the endangered giant sable antelope (Hippotragus niger variani): Insights into conservation and taxonomy. Mol. Phylogenet. Evol. 2015, 83, 242–249. [Google Scholar] [CrossRef]
- Węcek, K.; Hartmann, S.; Paijmans, J.L.A.; Taron, U.; Xenikoudakis, G.; Cahill, J.A.; Heintzman, P.D.; Shapiro, B.; Baryshnikov, G.; Bunevich, A.N.; et al. Complex admixture preceded and followed the extinction of wisent in the wild. Mol. Biol. Evol. 2017, 34, 598–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCesare, N.J.; Weckworth, B.V.; Pilgrim, K.L.; Walker, A.B.D.; Bergman, E.J.; Colson, K.E.; Corrigan, R.; Harris, R.B.; Hebblewhite, M.; Jesmer, B.R.; et al. Phylogeography of moose in western North America. J. Mammal. 2020, 101, 10–23. [Google Scholar] [CrossRef]
- Themudo, G.E.; Campos, P.F. Phylogenetic position of the extinct blue antelope, Hippotragus leucophaeus (Pallas, 1766) (Bovidae: Hippotraginae), based on complete mitochondrial genomes. Zool. J. Linn. Soc. 2018, 182, 225–235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Barido-Sottani, J.; Bošková, V.; Plessis, L.D.; Kühnert, D.; Magnus, C.; Mitov, V.; Müller, N.F.; PečErska, J.; Rasmussen, D.A.; Zhang, C.; et al. Taming the BEAST—A community teaching material resource for BEAST 2. Syst. Biol. 2018, 67, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Jia, F.; Lo, N.; Ho, S.Y.W. The impact of modelling rate heterogeneity among sites on phylogenetic estimates of intraspecific evolutionary rates and timescales. PLoS ONE 2014, 9, e95722. [Google Scholar] [CrossRef]
- Hopwood, A.T. New fossil mammals from Olduvai, Tanganyika Territory. Ann. Mag. Nat. Hist. 1934, 14, 546–550. [Google Scholar] [CrossRef]
- Gentry, A.W.; Gentry, A. Fossil Bovidae (Mammalia) of Olduvai Gorge, Tanzania, part 1. Bull. Br. Mus. Nat. Hist. Geol. 1978, 29, 289–446. [Google Scholar]
- Harris, J.M. Family Bovidae. In Koobi Fora Research Project Volume III; Harris, J.M., Ed.; Koobi Fora Research Project; Clarendon Press: Oxford, UK, 1991; Volume 3, pp. 139–320. [Google Scholar]
- Brink, J.S. Postcranial evidence for the evolution of the black wildebeest, Connochaetes gnou: An exploratory study. Palaeont. Afr. 1993, 30, 61–69. [Google Scholar]
- Vrba, E.S. New fossils of Alcelaphini and caprinae (Bovidae: Mammalia) from Awash, Ethiopia, and phylogenetic analysis of Alcelaphini. Palaeont. Afr. 1997, 34, 127–198. [Google Scholar]
- Arctander, P.; Johansen, C.; Coutellec-Vreto, M.-A. Phylogeography of three closely related African bovids (tribe Alcelaphini). Mol. Biol. Evol. 1999, 16, 1724–1739. [Google Scholar] [CrossRef] [Green Version]
- Brink, J.S. The Evolution of the Black Wildebeest, Connochaetes gnou, and Modern Large Mammal Faunas in Central Southern Africa. Ph.D. Thesis, University of Stellenbosch, Stellenbosch, South Africa, 2005. [Google Scholar]
- Bibi, F.; Rowan, J.; Reed, K. Late Pliocene Bovidae from Ledi-Geraru (Lower Awash Valley, Ethiopia) and their implications for Afar Paleoecology. J. Vertebr. Paleontol. 2017, 37, e1337639. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Solounias, N.; Barry, J.C.; Bernor, R.L.; Lindsay, E.H.; Raza, S.M. The oldest bovid from the Siwaliks, Pakistan. J. Vertebr. Paleontol. 1995, 15, 806–814. [Google Scholar] [CrossRef]
- Geraads, D.; Blondel, C.; Likius, A.; Mackaye, H.T.; Vignaud, P.; Brunet, M. New Hippotragini (Bovidae, mammalia) from the Late Miocene of Toros-Menalla (Chad). J. Vertebr. Paleontol. 2008, 28, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Lebatard, A.-E.; Bourlès, D.L.; Duringer, P.; Jolivet, M.; Braucher, R.; Carcaillet, J.; Schuster, M.; Arnaud, N.; Monié, P.; Lihoreau, F.; et al. Cosmogenic nuclide dating of Sahelanthropus tchadensis and Australopithecus bahrelghazali: Mio-Pliocene hominids from Chad. Proc. Natl. Acad. Sci. USA 2008, 105, 3226–3231. [Google Scholar] [CrossRef] [Green Version]
- Gentry, A.W. Fossil Bovidae (Mammalia) from Langebaanweg, South Africa. Ann. S. Afr. Mus. 1980, 79, 213–337. [Google Scholar]
- Hendey, Q.B. Geological succession at Langebaanweg, Cape Province, and global events of the Late Tertiary. S. Afr. J. Sci. 1981, 77, 33–38. [Google Scholar]
- Vrba, E.S.; Gatesy, J. New antelope fossils from Awash, Ethiopia, and phylogenetic analysis of Hippotragini (Bovidae, mammalia). Palaeont. Afr. 1994, 31, 55–72. [Google Scholar]
- Deino, A.L. 40Ar/39Ar dating of Laetoli, Tanzania. In Paleontology and Geology of Laetoli: Human Evolution in Context; Harrison, T., Ed.; Springer: New York, NY, USA, 2011; Volume 1, pp. 77–97. [Google Scholar]
- Gentry, A.W. Bovidae. In Paleontology and Geology of Laetoli: Human Evolution in Context; Harrison, T., Ed.; Springer: Dordrecht, The Nertherlands; Heidelberg, Germany; London, UK; New York, NY, USA, 2011; Volume 2, pp. 363–465. [Google Scholar]
- Hedrick, P.W. Conservation genetics and North American bison (Bison bison). J. Hered. 2009, 100, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, J.M.; Kawałko, A.; Tokarska, M.; Jaarola, M.; Vallenback, P.; Pertoldi, C. Post-bottleneck mtDNA diversity in a free-living population of European bison: Implications for conservation. J. Zool. 2009, 277, 81–87. [Google Scholar] [CrossRef]
- Lott, D.F. American Bison: A Natural History; Organisms and Environments; University of California Press: Berkley, CA, USA; Los Angeles, CA, USA; London, UK, 2002. [Google Scholar]
- IUCN SSC Antelope Specialist Group. Oryx dammah. IUCN Red List Threat. Species 2016, e.T15568A50191470. [Google Scholar] [CrossRef]
- Bouman, I.; Bouman, J. The history of Prewalski’s horse. In Przewalski’s Horse—The History and Biology of an Endangered Species; Boyd, L., Houpt, K.A., Eds.; State University of New York Press: Albany, NY, USA, 1994; pp. 5–38. [Google Scholar]
- Bazin, E.; Glémin, S.; Galtier, N. Population size does not influence mitochondrial genetic diversity in animals. Science 2006, 312, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Drummond, A.J.; Rambaut, A.; Wilson, M.C.; Matheus, P.E.; Sher, A.V.; Pybus, O.G.; Gilbert, M.T.P.; Barnes, I.; Binladen, J.; et al. Rise and fall of the Beringian steppe bison. Science 2004, 306, 1561–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, E.D.; Heller, R.; Siegismund, H.R. Comparative phylogeography of African savannah ungulates. Mol. Ecol. 2012, 21, 3656–3670. [Google Scholar] [CrossRef] [PubMed]
- Lerp, H.; Wronski, T.; Pfenninger, M.; Plath, M. A phylogeographic framework for the conservation of Saharan and Arabian dorcas gazelles (Artiodactyla: Bovidae). Org. Divers. Evol. 2011, 11, 317–329. [Google Scholar] [CrossRef]
- Senn, H.; Banfield, L.; Wacher, T.; Newby, J.; Rabeil, T.; Kaden, J.; Kitchener, A.C.; Abaigar, T.; Silva, T.L.; Maunder, M.; et al. Splitting or lumping? A conservation dilemma exemplified by the critically endangered dama gazelle (Nanger dama). PLoS ONE 2014, 9, e98693. [Google Scholar] [CrossRef] [Green Version]
- Sicks, F.; Tierpark Berlin, Berlin, Germany. Personal communication, 2021.
- Kingdon, J. Genus Addax. In Mammals of Africa; Bloomsbury: London, UK; New Dehli, India; New York, NY, USA; Sydney, Australia, 2013; Volume 6, p. 566. [Google Scholar]
- deMenocal, P.B. Plio-Pleistocene African climate. Science 1995, 270, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L. Orbital scale vegetation change in Africa. Quat. Sci. Rev. 2011, 30, 3589–3602. [Google Scholar] [CrossRef]
- Skonieczny, C.; McGee, D.; Winckler, G.; Bory, A.; Bradtmiller, L.I.; Kinsley, C.W.; Polissar, P.J.; De Pol-Holz, R.; Rossignol, L.; Malaizé, B. Monsoon-driven Saharan dust variability over the past 240,000 years. Sci. Adv. 2019, 5, eaav1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.S.; Stone, J.R.; Beuning, K.R.M.; Park, L.E.; Reinthal, P.N.; Dettman, D.; Scholz, C.A.; Johnson, T.C.; King, J.W.; Talbot, M.R.; et al. Ecological consequences of early Late Pleistocene megadroughts in tropical Africa. Proc. Natl. Acad. Sci. USA 2007, 104, 16422–16427. [Google Scholar] [CrossRef] [Green Version]
- Scholz, C.A.; Johnson, T.C.; Cohen, A.S.; King, J.W.; Peck, J.A.; Overpeck, J.T.; Talbot, M.R.; Brown, E.T.; Kalindekafe, L.; Amoako, P.Y.O.; et al. East African megadroughts between 135 and 75 thousand years ago and bearing on early-modern human origins. Proc. Natl. Acad. Sci. USA 2007, 104, 16416–16421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, N.A.; El-Hawat, A.S.; Turner, P.; Armitage, S.J.; Salem, M.J.; White, K.H.; McLaren, S. Palaeohydrology of the Fazzan Basin and surrounding regions: The last 7 million years. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2008, 263, 131–145. [Google Scholar] [CrossRef]
- Osborne, A.H.; Vance, D.; Rohling, E.J.; Barton, N.; Rogerson, M.; Fello, N. A humid corridor across the Sahara for the migration of early modern humans out of Africa 120,000 years ago. Proc. Natl. Acad. Sci. USA 2008, 105, 16444–16447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrasoaña, J.C.; Roberts, A.P.; Rohling, E.J. Dynamics of green Sahara Periods and their role in hominin evolution. PLoS ONE 2013, 8, e76514. [Google Scholar] [CrossRef] [Green Version]
- Morrow, C.; Molcanova, R.; Wacher, T. Oryx dammah Scimitar-horned oryx (scimitar oryx). In Mammals of Africa; Bloomsbury: London, UK; New Dehli, India; New York, NY, USA; Sydney, Australia, 2013; Volume 6, pp. 586–592. [Google Scholar]
- Landers, J. Rewilding the African Scimitar-Horned Oryx. Available online: https://www.smithsonianmag.com/smithsonian-institution/rewilding-african-scimitar-horned-oryx-180958796/ (accessed on 7 January 2021).
- Cassinello, J.; Cuzin, F.; Jdeidi, T.; Masseti, M.; Nader, I.; de Smet, K. Ammotragus lervia. IUCN Red List Threat. Species 2008, e.T1151A3288917. [Google Scholar] [CrossRef]
- IUCN SSC Antelope Specialist Group. Gazella leptoceros. IUCN Red List Threat. Species 2016, e.T8972A50186909. [Google Scholar] [CrossRef]
- Newby, J. Saving the addax. Oryx 2006, 40, 256. [Google Scholar] [CrossRef] [Green Version]
- Brüniche-Olsen, A.; Kellner, K.F.; Anderson, C.J.; DeWoody, J.A. Runs of homozygosity have utility in mammalian conservation and evolutionary studies. Conserv. Genet. 2018, 19, 1295–1307. [Google Scholar] [CrossRef]
- Riordan, P.; Gilbert, T.; Petretto, M.; Senn, H.; Craig, M.S.; Banfield, L.M.; Ivy, J.; Abid, H.; Nouioui, M. The Genetics of Reintroduced Addax Populations in Tunisia: Moving towards a Global Management Plan; Year 1 Project Report; Marwell Wildlife: Winchester, UK, 2018. [Google Scholar]
- Krause, F.; Zoo Hannover, Hannover, Germany. Personal communication, 2021.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Country | Location | Collector (s) | Collection Date | Sample Type | Accession Number Mitochondrial Genome |
|---|---|---|---|---|---|---|
| ZMB MAM 2165 | Sudan | Sennar | Hemprich and Ehrenberg | 1821 | bone & skin | MZ474955 |
| ZMB MAM 2166 | Sudan | Sennar | Hemprich and Ehrenberg | 1821 | skin | MZ474956 |
| ZMB MAM 2167 | Sudan | Sennar | Hemprich and Ehrenberg | 1821 | skin | MZ474957 |
| ZMB MAM 35370 | Western Sahara | Dakhla | Spatz | 1926 | bone | MZ474958 |
| ZMB MAM 7424 | Tunisia | Tunis, Gabès | Spatz | 1884–1903(?) | bone | MZ474959 |
| ZMB MAM 8836 | Tunisia | Tunis | Spatz | 1884–1903(?) | bone | MZ474960 |
| ZMB MAM 8837 | Libya | Tripoli | Browski | 1895–? | bone | MZ474961 |
| ZMB MAM 8838 | Libya | Tripoli | Browski | 1895–? | bone | MZ474962 |
| ZMB MAM 8839 | Libya | Tripoli | Browski | 1895–? | bone | MZ474963 |
| ZMB MAM 8840 | Libya | Tripoli | Browski | 1895–? | bone | MZ474964 |
| IZW 607/10 | Germany | Tierpark Berlin | - | - | liver | MZ474965 |
| Calibration Point | Prior Type | Age [Mya] | 95% Range [Mya] | Fossil Taxon | Site/Geological Unit | References |
|---|---|---|---|---|---|---|
| Crown Bovidae | Normal | 18 | 16.0–20.0 | Eotragus noyei | Kamlial and Vihowa Formations, Pakistan | [108] |
| Stem Hippotragini | Log normal | 6.4 | 6.4–8 | Saheloryx tchadensis, Saheloryx solidus, Tchadotragus sudrei | Anthracotheriid unit at Toros-Menalla, Chad | [109,110] |
| Crown Alcelaphini | Log normal | 4.5 | 4.5–5.625 | Damalacra neanica | Pelletal Phosphorite Member at Langebaanweg, South Africa | [103,111,112] |
| Crown Hippotragini | Log normal | 3.6 | 3.6–4.5 | Hippotragus sp., Oryx sp. | Lower Laetoli Beds at Laetoli, Tanzania | [113,114,115] |
| Crown Connochaetes spp. | Log normal | 1.0 | 1.0–1.25 | Connochaetes gnou | Cornelia-Uitzoek, South Africa | [102,105,106] |
| Total Assembly Length | 2,795,176,578 bp |
| Number of Scaffolds | 86,926 |
| Scaffold N50 | 20,757,513 |
| Scaffold L50 | 37 |
| Longest Scaffold | 87,765,150 bp |
| GC Content | 41.72% |
| Cetartiodactyla BUSCO Scores | Laurasiatheria BUSCO Scores | Mammalia BUSCO Scores | |
|---|---|---|---|
| C | 91.6% | 92.4% | 91.2% |
| Complete BUSCOs | 12,209 | 11,312 | 8418 |
| Complete and Single-Copy BUSCOs | 12,038 | 11,151 | 8204 |
| Complete and Duplicated BUSCOs | 171 | 161 | 114 |
| Fragmented BUSCOs | 341 | 278 | 287 |
| Missing BUSCOs | 785 | 644 | 521 |
| Total BUSCO Groups Searched | 13,335 | 12,234 | 9226 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hempel, E.; Westbury, M.V.; Grau, J.H.; Trinks, A.; Paijmans, J.L.A.; Kliver, S.; Barlow, A.; Mayer, F.; Müller, J.; Chen, L.; et al. Diversity and Paleodemography of the Addax (Addax nasomaculatus), a Saharan Antelope on the Verge of Extinction. Genes 2021, 12, 1236. https://doi.org/10.3390/genes12081236
Hempel E, Westbury MV, Grau JH, Trinks A, Paijmans JLA, Kliver S, Barlow A, Mayer F, Müller J, Chen L, et al. Diversity and Paleodemography of the Addax (Addax nasomaculatus), a Saharan Antelope on the Verge of Extinction. Genes. 2021; 12(8):1236. https://doi.org/10.3390/genes12081236
Chicago/Turabian StyleHempel, Elisabeth, Michael V. Westbury, José H. Grau, Alexandra Trinks, Johanna L. A. Paijmans, Sergei Kliver, Axel Barlow, Frieder Mayer, Johannes Müller, Lei Chen, and et al. 2021. "Diversity and Paleodemography of the Addax (Addax nasomaculatus), a Saharan Antelope on the Verge of Extinction" Genes 12, no. 8: 1236. https://doi.org/10.3390/genes12081236
APA StyleHempel, E., Westbury, M. V., Grau, J. H., Trinks, A., Paijmans, J. L. A., Kliver, S., Barlow, A., Mayer, F., Müller, J., Chen, L., Koepfli, K. -P., Hofreiter, M., & Bibi, F. (2021). Diversity and Paleodemography of the Addax (Addax nasomaculatus), a Saharan Antelope on the Verge of Extinction. Genes, 12(8), 1236. https://doi.org/10.3390/genes12081236