
Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients


 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
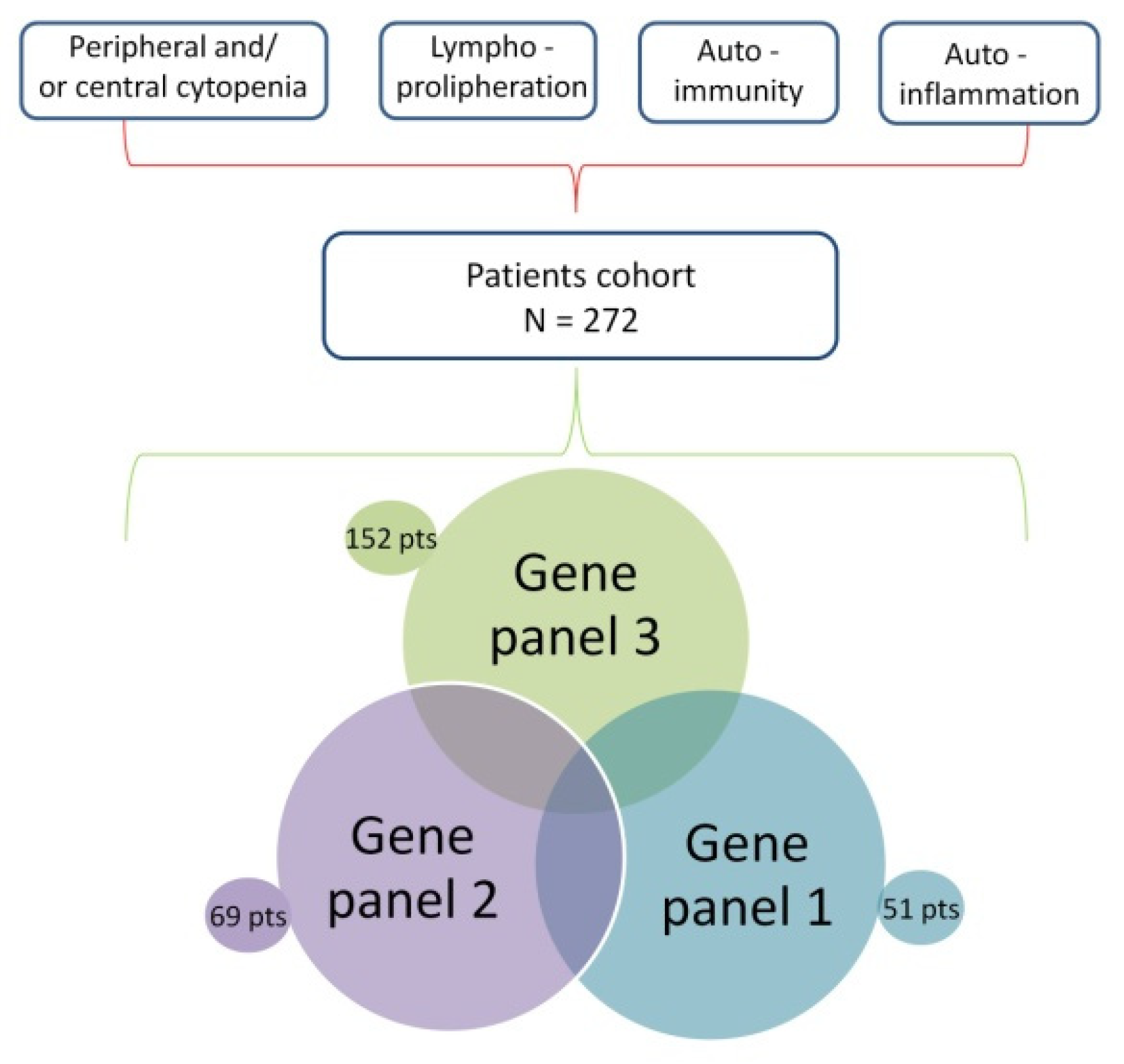
2.1. Patient Recruitment
2.2. Study Design
2.3. Library Design and Sequencing, Bioinformatic Analysis, and Sanger Validation
3. Results
3.1. Pathogenic or likely Pathogenic Variants Detected
3.2. Variants of Unknown Significance with a Probable Effect on the Phenotype
3.3. Wide Genetic Variability in Immune Dysregulation Disorders: Low-Impact Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, V.; Dotta, L.; Giardino, G.; Cirillo, E.; Lougaris, V.; D’Assante, R.; Prandini, A.; Consolini, R.; Farrow, E.G.; Thiffault, I.; et al. Diagnostics of Primary Immunodeficiencies through Next-Generation Sequencing. Front. Immunol. 2016, 7, 466. [Google Scholar] [CrossRef] [Green Version]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity with Immune Dysregulation: From Bench to Bedside. Front. Pediatr. 2019, 7, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.Y.; Leiding, J.W.; Liu, X.; Logan, B.R.; Burroughs, L.M.; Allenspach, E.J.; Skoda-Smith, S.; Uzel, G.; Notarangelo, L.D.; Slatter, M.; et al. Hematopoietic Cell Transplantation in Patients With Primary Immune Regulatory Disorders (PIRD): A Primary Immune Deficiency Treatment Consortium (PIDTC) Survey. Front. Immunol. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCusker, C.; Upton, J.; Warrington, R. Primary immunodeficiency. Allergy Asthma Clin. Immunol. 2018, 14, 1–12. [Google Scholar] [CrossRef]
- Yamashita, M.; Inoue, K.; Okano, T.; Morio, T. Inborn errors of immunity—Recent advances in research on the pathogenesis. Inflamm. Regen. 2021, 41, 1–7. [Google Scholar] [CrossRef]
- Shield, A.M.; Patel, S.Y. The primary immunodeficiency disorders. Medicine 2018, 45, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Seidel, M.G. Treatment of immune-mediated cytopenias in patients with primary immunodeficiencies and immune regulatory disorders (PIRDs). Hematology 2020, 2020, 673–679. [Google Scholar] [CrossRef]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; Van Montfrans, J.; Scheible, R.; Rusch, S.; Gasteiger, L.M.; Grimbacher, B.; et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pr. 2019, 7, 1763–1770. [Google Scholar] [CrossRef]
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35–e40. [Google Scholar] [CrossRef]
- Oliveira, J.B. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr. Opin. Pediatr. 2013, 25, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Seleman, M.; Hoyos-Bachiloglu, R.; Geha, R.S.; Chou, J. Uses of Next-Generation Sequencing Technologies for the Diagnosis of Primary Immunodeficiencies. Front. Immunol. 2017, 8, 847. [Google Scholar] [CrossRef] [Green Version]
- Al-Mousa, H.; Abouelhoda, M.; Monies, D.M.; Al Tassan, N.; Al-Ghonaium, A.; Al-Saud, B.; Al-Dhekri, H.; Arnaout, R.; Al-Muhsen, S.; Ades, N.; et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J. Allergy Clin. Immunol. 2016, 137, 1780–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisgin, A.; Boga, I.; Yilmaz, M.; Bingol, G.; Altintas, D. The Utility of Next-Generation Sequencing for Primary Immunodeficiency Disorders: Experience from a Clinical Diagnostic Laboratory. BioMed Res. Int. 2018, 2018, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, C.; Bogunovic, D. Incomplete penetrance in primary immunodeficiency: A skeleton in the closet. Qual. Life Res. 2020, 139, 745–757. [Google Scholar] [CrossRef]
- Byun, M.; Abhyankar, A.; Lelarge, V.; Plancoulaine, S.; Palanduz, A.; Telhan, L.; Boisson, B.; Picard, C.; Dewell, S.; Zhao, C.; et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010, 207, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijman, I.; van Montfrans, J.M.; Hoogstraat, M.; Boes, M.L.; van de Corput, L.; Renner, E.D.; van Zon, P.; van Lieshout, S.; Elferink, M.G.; van der Burg, M.; et al. Targeted next-generation sequencing: A novel diagnostic tool for primary immunodeficiencies. J. Allergy Clin. Immunol. 2014, 133, 529–534.e1. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, J.L.; Niemela, J.E.; Fleisher, T.A.; Rosenzweig, S.D. Targeted NGS: A Cost-Effective Approach to Molecular Diagnosis of PIDs. Front. Immunol. 2014, 5, 531. [Google Scholar] [CrossRef] [Green Version]
- Picard, C.; Gaspar, H.B.; Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2017, 38, 96–128. [Google Scholar] [CrossRef] [Green Version]
- Ceccherini, I.; Rusmini, M.; Arostegui, J.I. Genetic Aspects of Investigating and Understanding Autoinflammation. In Textbook of Autoinflammation, 1st ed.; Hashkes, P.J., Laxer, R.M., Simon, A., Eds.; Springer Nature: Basingstoke, UK, 2019; pp. 19–48. [Google Scholar]
- Farmer, J.R.; Mahajan, V.S. Molecular Diagnosis of Inherited Immune Disorders. Clin. Lab. Med. 2019, 39, 685–697. [Google Scholar] [CrossRef]
- French, M.; Tangye, S.G. The Next Generation of Diagnostic Tests for Primary Immunodeficiency Disorders. J. Infect. Dis. 2019, 221, 1232–1234. [Google Scholar] [CrossRef]
- Rudilla, F.; Franco-Jarava, C.; Gallo, M.M.; Garcia-Prat, M.; Martín-Nalda, A.; Rivière, J.G.; Aguiló-Cucurull, A.; Mongay, L.; Vidal, F.; Solanich, X.; et al. Expanding the Clinical and Genetic Spectra of Primary Immunodeficiency-Related Disorders with Clinical Exome Sequencing: Expected and Unexpected Findings. Front. Immunol. 2019, 10, 2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinn, I.K.; Chan, A.Y.; Chen, K.; Chou, J.; Dorsey, M.J.; Hajjar, J.; Jongco, A.M.; Keller, M.D.; Kobrynski, L.J.; Kumanovics, A.; et al. Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2019, 145, 46–69. [Google Scholar] [CrossRef] [Green Version]
- Leavis, H.; Zwerina, J.; Manger, B.; Fritsch-Stork, R.D.E. Novel Developments in Primary Immunodeficiencies (PID)—A Rheumatological Perspective. Curr. Rheumatol. Rep. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Yska, H.A.F.; Elsink, K.; Kuijpers, T.W.; Frederix, G.W.J.; Van Gijn, M.E.; Van Montfrans, J.M. Diagnostic Yield of Next Generation Sequencing in Genetically Undiagnosed Patients with Primary Immunodeficiencies: A Systematic Review. J. Clin. Immunol. 2019, 39, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Hoyos-Bachiloglu, R.; Chou, J. Autoimmunity and immunodeficiency. Curr. Opin. Rheumatol. 2020, 32, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Heimall, J.R.; Hagin, D.; Hajjar, J.; Henrickson, S.; Hernandez-Trujillo, H.S.; Tan, Y.; Kobrynski, L.; Paris, K.; Torgerson, T.R.; Verbsky, J.W.; et al. Use of Genetic Testing for Primary Immunodeficiency Patients. J. Clin. Immunol. 2018, 38, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Takafuji, S.; Mori, T.; Nishimura, N.; Yamamoto, N.; Uemura, S.; Nozu, K.; Terui, K.; Toki, T.; Ito, E.; Muramatsu, H.; et al. Usefulness of functional splicing analysis to confirm precise disease pathogenesis in Diamond-Blackfan anemia caused by intronic variants in RPS19. Pediatr. Hematol. Oncol. 2021, 1–16. [Google Scholar] [CrossRef]
- Rusmini, M.; Federici, S.; Caroli, F.; Grossi, A.; Baldi, M.; Obici, L.; Insalaco, A.; Tommasini, A.; Caorsi, R.; Gallo, E.; et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann. Rheum. Dis. 2015, 75, 1550–1557. [Google Scholar] [CrossRef]
- Papa, R.; Rusmini, M.; Volpi, S.; Caorsi, R.; Picco, P.; Grossi, A.; Caroli, F.; Bovis, F.; Musso, V.; Obici, L.; et al. Next generation sequencing panel in undifferentiated autoinflammatory diseases identifies patients with colchicine-responder recurrent fevers. Rheumatology 2019, 59, 344–360. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Trotta, L.; Martelius, T.; Siitonen, T.; Hautala, T.; Hämäläinen, S.; Juntti, H.; Taskinen, M.; Ilander, M.; Andersson, E.I.; Zavialov, A.; et al. ADA2 deficiency: Clonal lymphoproliferation in a subset of patients. J. Allergy Clin. Immunol. 2018, 141, 1534–1537.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miano, M.; Grossi, A.; Dell’Orso, G.; Lanciotti, M.; Fioredda, F.; Palmisani, E.; Lanza, T.; Guardo, D.; Beccaria, A.; Ravera, S.; et al. Genetic screening of children with marrow failure. The role of primary Immunodeficiencies. Am. J. Hematol. 2021, 96, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Alsultan, A.; Basher, E.; Alqanatish, J.; Mohammed, R.; Alfadhel, M. Deficiency of ADA2 mimicking autoimmune lymphoproliferative syndrome in the absence of livedo reticularis and vasculitis. Pediatr. Blood Cancer 2017, 65, e26912. [Google Scholar] [CrossRef]
- Westermann-Clark, E.; Grossi, A.; Fioredda, F.; Giardino, S.; Cappelli, E.; Terranova, P.; Palmisani, E.; Farmer, J.; Foldvari, Z.; Yamazaki, Y.; et al. RAG deficiency with ALPS features successfully treated with TCRαβ/CD19 cell depleted haploidentical stem cell transplant. Clin. Immunol. 2017, 187, 102–103. [Google Scholar] [CrossRef] [Green Version]
- Maggiore, R.; Grossi, A.; Fioredda, F.; Palmisani, E.; Terranova, P.; Cappelli, E.; Lanza, T.; Pierri, F.; Guardo, D.; Calvillo, M.; et al. Unusual Late-onset Enteropathy in a Patient with Lipopolysaccharide-responsive Beige-like Anchor Protein Deficiency. J. Pediatr. Hematol. 2019, 42, e768–e771. [Google Scholar] [CrossRef]
- Fioredda, F.; Rotulo, G.A.; Farruggia, P.; Dagliano, F.; Pillon, M.; Trizzino, A.; Notarangelo, L.; Luti, L.; Lanza, T.; Terranova, P.; et al. Late-onset and long-lasting autoimmune neutropenia: An analysis from the Italian Neutropenia Registry. Blood Adv. 2020, 4, 5644–5649. [Google Scholar] [CrossRef]
- Witzel-Schlömp, K.; Hobart, M.J.; Fernie, B.A.; Orren, A.; Würzner, R.; Rittner, C.; Kaufmann, T.; Schneider, P.M. Heterogeneity in the genetic basis of human complement C9 deficiency. Immunogenetics 1998, 48, 144–147. [Google Scholar] [CrossRef]
- Pan-Hammarström, Q.; Salzer, U.; Du, L.; Björkander, J.; Cunningham-Rundles, C.; Nelson, D.L.; Bacchelli, C.; Gaspar, H.B.; Offer, S.; Behrens, T.W.; et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 2007, 39, 429–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.S.; Recher, M.; Giliani, S.; Walter, J.E.; Lee, Y.N.; Frugoni, F.; Maddox, D.; Kirmani, S.; Notarangelo, L.D. Adult-onset manifestation of idiopathic T-cell lymphopenia due to a heterozygous RAG1 mutation. J. Allergy Clin. Immunol. 2013, 131, 1421–1423. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.E.; Lo, M.S.; Kis-Toth, K.; Tirosh, I.; Frugoni, F.; Lee, Y.N.; Csomos, K.; Chen, K.; Pillai, S.; Dunham, J.; et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J. Allergy Clin. Immunol. 2014, 135, 272–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasek, M.; Bojarska-Junak, A.; Wagner, M.; Sobczyński, M.; Wołowiec, D.; Rolinski, J.M.; Karabon, L.; Kuśnierczyk, P. Association of variants in BAFF (rs9514828 and rs1041569) and BAFF-R (rs61756766) genes with the risk of chronic lymphocytic leukemia. Tumor Biol. 2016, 37, 13617–13626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miano, M.; Cappelli, E.; Pezzulla, A.; Venè, R.; Grossi, A.; Terranova, P.; Palmisani, E.; Maggiore, R.; Guardo, D.; Lanza, T.; et al. FAS-mediated apoptosis impairment in patients with ALPS/ALPS-like phenotype carrying variants on CASP10 gene. Br. J. Haematol. 2019, 187, 502–508. [Google Scholar] [CrossRef]
- Fioredda, F.; Cappelli, E.; Mariani, A.; Beccaria, A.; Palmisani, E.; Grossi, A.; Ceccherini, I.; Venè, R.; Micalizzi, C.; Calvillo, M.; et al. Thrombotic thrombocytopenic purpura and defective apoptosis due to CASP8/10 mutations: The role of mycophenolate mofetil. Blood Adv. 2019, 3, 3432–3435. [Google Scholar] [CrossRef]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Cifaldi, C.; Brigida, I.; Barzaghi, F.; Zoccolillo, M.; Ferradini, V.; Petricone, D.; Cicalese, M.P.; Lazarevic, D.; Cittaro, D.; Omrani, M.; et al. Corrigendum: Targeted NGS Platforms for Genetic Screening and Gene Discovery in Primary Immunodeficiencies. Front. Immunol. 2019, 10, 1184. [Google Scholar] [CrossRef] [Green Version]
- Almarzooqi, F.; Souid, A.-K.; Vijayan, R.; Al-Hammadi, S. Novel genetic variants of inborn errors of immunity. PLoS ONE 2021, 16, e0245888. [Google Scholar] [CrossRef]
- Bisgin, A.; Sonmezler, O.; Boga, I.; Yilmaz, M. The impact of rare and low-frequency genetic variants in common variable immunodeficiency (CVID). Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Mazzolari, E.; Moshous, D.; Forino, C.; De Martiis, D.; Offer, C.; Lanfranchi, A.; Giliani, S.; Imberti, L.; Pasic, S.; Ugazio, A.G.; et al. Hematopoietic stem cell transplantation in Omenn syndrome: A single-center experience. Bone Marrow Transplant. 2005, 36, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Hadjadj, J.; Aladjidi, N.; Fernandes, H.; Leverger, G.; Magérus-Chatinet, A.; Mazerolles, F.; Stolzenberg, M.-C.; Jacques, S.; Picard, C.; Rosain, J.; et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood 2019, 134, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Rachid, R.; Castigli, E.; Geha, R.S.; Bonilla, F.A. TACI mutation in common variable immunodeficiency and IgA deficiency. Curr. Allergy Asthma Rep. 2006, 6, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; van Oostrom, D.; Li, J.; Savelkoul, H.F.J. Innate Mechanisms in Selective IgA Deficiency. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Gallo, M.M.; Radigan, L.; Almejún, M.B.; Martínez-Pomar, N.; Matamoros, N.; Cunningham-Rundles, C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J. Allergy Clin. Immunol. 2013, 131, 468–476. [Google Scholar] [CrossRef]
- Salzer, U.; Bacchelli, C.; Buckridge, S.; Pan-Hammarström, Q.; Jennings, S.; Lougaris, V.; Bergbreiter, A.; Hagena, T.; Birmelin, J.; Plebani, A.; et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 2009, 113, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Arts, P.; Simons, A.; Alzahrani, M.S.; Yilmaz, E.; Alidrissi, E.; Van Aerde, K.J.; Alenezi, N.; Alghamdi, H.A.; Aljubab, H.A.; Al-Hussaini, A.A.; et al. Exome sequencing in routine diagnostics: A generic test for 254 patients with primary immunodeficiencies. Genome Med. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Grossi, A.; Cusano, R.; Rusmini, M.; Penco, F.; Schena, F.; Podda, R.A.; Caorsi, R.; Gattorno, M.; Uva, P.; Ceccherini, I. ADA2 deficiency due to a novel structural variation in 22q11.1. Clin. Genet. 2019, 95, 732–733. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ID | GENDER | Total Variants Called | Filtered Variants ‡ | Gene | Inherit. of Associated Phenotype | Variants § | ClinVar | Zygosity | Variant Classific. * | dbSNP (#rs) | CADD Score | Frequency (gnomAD) | DANN Score | FATHMM | SIFT | PROVEAN | Cases with Same Variant (#) | Parental Inheritance/de novo | Previous Genetic Tests | Clinical Phenotype | Consensus *** |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | F | 592 | 17 | TNFRSF13B | AD/AR | p.C104R | conflicting | HET | P | 34557412 | 25.8 | 3.92 × 10−3 | 0.9172 | D | D | D | 4 | M | ALPS-like | 1 | |
| 10 | F | 1335 | 21 | C9 | - | p.C125 * | - | HET | LP | na | 32 | - | 0.985 | - | - | - | 1 | M | ALPS-like | 4 | |
| 15 | M | 516 | 16 | RAG1 | AR | p.R507Q | - | HOMO | LP | 143969029 | 28.7 | 6.57 × 10−6 | 0.9994 | T | D | D | 1 | M, F | ALPS-like | 1 | |
| 19 | F | 497 | 14 | IKBKG | XLR | p.E125K | B/LB | HET | LP | 148695964 | 28 | 1.50 × 10−3 | 0.9991 | D | D | D | 2 | M WT F na | ALPS-like | 2 | |
| 22 | M | 528 | 12 | TNFRSF13B | AD/AR | p.C193 * | conflicting | HET | LP | 72553885 | 35 | 5.68 × 10−5 | 0.985 | - | - | - | 2 | F | ALPS-like | 1 | |
| 32 | F | 594 | 11 | STAT3 | AD | p.K658R | LP | HET | LP | na | 25.5 | - | 0.999 | D | T | N | 1 | Somatic | ALPS-like | 1 | |
| ADA2 | AR | p.L188P | uncertain significance | HET | LP | 760102576 | 26.8 | 1.97 × 10−5 | 0.999 | D | D | D | 1 | M | 1 | ||||||
| ADA2 | AR | p.T187P | - | HET | LP | 752890414 | 26.3 | 3.99 × 10−6 | 0.996 | T | D | D | 1 | F | |||||||
| 35 | F | 640 | 13 | CTLA4 | AD | p.C58S fs*13 | P | HET | LP | na | - | - | 0.991 | - | - | - | 1 | M | Sanger FAS | ALPS-like | 1 |
| 39 | M | 661 | 8 | LRBA | AR | p.R655 * | - | HOMO | P | 199750191 | 42 | 6.58 × 10−6 | 0.998 | - | - | - | 1 | M, F | ALPS-like | 1 | |
| 51 | F | 655 | 11 | ELANE | AD | c.597 +1G>A | P | HET | P | 1555710005 | 26.7 | - | 0.907 | - | - | - | 1 | na | Neutropenia | 1 | |
| 64 | F | 1928 | 33 | RPS19 | AD | p.R62W | P | HET | P | 104894711 | 24.9 | - | 0.999 | D | - | - | 1 | M | BMF | 1 | |
| 66 | M | 683 | 10 | SMARCAL1 | AR | p.R499W | - | HET | LP | 1302790588 | 25.3 | 3.98 × 10−6 | 0.9987 | D | D | D | 1 | na | ALPS-like | 4 | |
| 75 | F | 718 | 12 | RAG1 | AR | p.Q407E | LP | HET | LP | na | 25.1 | - | 0.986 | T | D | N | 1 | M | Sanger ELANE | ALPS-like | 2 |
| 80 | M | 1225 | 14 | IKBKG | XLR | p.E125K | B/LB | HEMIZIG | LP | 148695964 | 28 | 1.50 × 10−3 | 0.9991 | D | D | D | 2 | M | ALPS-like | 1 | |
| 86 | M | 1470 | 21 | C8B | AR | p.R428 * | P | HET | P | 41286844 | 41 | 3.98 × 10−6 | 0.9984 | - | - | - | 1 | na | BMF | 4 | |
| FAN1 | AR | p.M86G fs*14 | - | HET | LP | 758406790 | - | 1.19 × 10−5 | - | - | - | - | 1 | na | 2 | ||||||
| 88 | F | 1386 | 22 | TNFRSF13B | AD/AR | p.C104Y | LP | HET | LP | 72553879 | 24.7 | 1.58 × 10−4 | 0.7764 | D | D | D | 2 | na | ALPS-like | 1 | |
| 90 | M | 1105 | 16 | NHEJ1 | - | p.R57 * | P | HET | P | 118204451 | 37 | 7.95 × 10−6 | 0.997 | - | - | - | 1 | na | ALPS-like | 1 | |
| 92 | M | 1196 | 18 | C7 | - | R521S | P | HET | LP | 121964920 | 22.3 | 2.35 × 10−3 | 0.9973 | T | D | D | 1 | F | ALPS-like | 1 | |
| 93 | M | 131 | 3 | TNFRSF13B | AD/AR | p.C193 * | conflicting | HET | LP | 72553885 | 36 | 3.99 × 10−6 | 0.985 | - | - | - | 2 | na | ALPS-like | 1 | |
| 94 | F | 1292 | 22 | NCF1 | AR | p.W193 * | P | HOMO | P | 145360423 | 36 | 5.53 × 10−4 | 0.995 | D | - | - | 1 | M, F | Immune-deficiency | 1 | |
| 97 | M | 815 | 28 | IL7R | AR | p.C118Y | P | HOMO | LP | 193922641 | 19.9 | 3.95 × 10−5 | 0.9369 | T | T | D | 1 | na | Sanger TERC, TERT | Immune-deficiency | 1 |
| 100 | F | 1614 | 22 | AIRE | AR | p.E517 * | - | HET | P | na | 48 | - | 0.994 | - | - | - | 1 | F | ALPS-like | 4 | |
| 105 | F | 1517 | 26 | AIRE | AR | p.R9W | LP | HET | LP | na | 23.6 | - | 0.998 | D | D | D | 1 | na | Sanger FAS | ALPS-like | 4 |
| 106 | F | 703 | 25 | RNASEH2B | AR | p.A177T | P/LP | HET | P | 75184679 | 24 | 1.45 × 10−3 | 0.9967 | D | T | N | 1 | na | BMF | 2 | |
| 109 | F | 1603 | 26 | TMEM173 | AD | p.V155M | P | HET | P | 587777610 | 24.7 | 2.63 × 10−5 | 0.999 | T | D | N | 1 | na | SAID | 1 | |
| 113 | F | 1780 | 35 | CASP8 | AR | p.R494 * | - | HET | P | 1368296717 | 37 | 3.98 × 10−6 | 0.996 | - | - | - | 1 | na | Sanger TERC | Immune-deficiency | 2 |
| 114 | F | 1123 | 10 | TNFRSF13B | AD/AR | p.L69T fs*12 | conflicting | HET | LP | 72553875 | 22.8 | 3.09 × 10−4 | - | - | - | - | 3 | na | ALPS-like | 1 | |
| 120 | M | 1736 | 33 | TNFRSF13B | AD/AR | p.S194 * | P | HET | P | 121908379 | 36 | - | - | - | - | - | 1 | na | ALPS-like | 1 | |
| 131 | M | 1450 | 25 | STAT3 | AD | p.R152W | P | HET | LP | 869312890 | 25.7 | 0.00 | 0.998 | T | D | D | 1 | na | Sanger FAS | ALPS-like | 1 |
| 135 | M | 1095 | 28 | SH3BP2 | AD | p.T531I | - | HET | LP | 746860671 | 21.7 | 3.98 × 10−6 | 0.9927 | T | D | N | 1 | na | ALPS | 3 | |
| 139 | M | 174 | 11 | SBDS | AR | c.258+2T>C | P | HET | P | 113993993 | 33 | 3.88 × 10−3 | - | - | - | - | 2 | na | Sanger FAS, ADA2 | ALPS-like | 2 |
| 162 | M | 106 | 4 | TNFRSF13B | AD/AR | p.C104R | conflicting | HET | P | 34557412 | 25.8 | 3.92 × 10−3 | 0.9172 | D | D | D | 4 | na | Sanger TERC, TINF2 | BMF | 1 |
| 178 | M | 111 | 5 | SBDS | AR | c.25 +2T>C | P | HET | P | 113993993 | 33 | 3.88 × 10−3 | - | - | - | - | 2 | na | Hystocytosis | 4 | |
| 182 | M | 169 | 11 | RAB27A | AR | p.I181M | uncertain significance | HET | LP | 139025012 | 17.7 | 9.19 × 10−5 | 0.9953 | T | D | N | 1 | na | AIHA | 4 | |
| 192 | F | 129 | 3 | FAS | AD | c.650_651+3del CTGTA insAGTG | uncertain significance | HET | LP | na | 14.95 | 3.98 × 10−6 | 0.8238 | - | - | - | 1 | na | ALPS-like | 1 | |
| 203 | F | 140 | 6 | TNFRSF13B | AD/AR | p.L69T fs*12 | conflicting | HET | LP | 72553875 | 22.8 | 3.09 × 10−4 | - | - | - | - | 3 | na | ALPS | 1 | |
| 206 | F | 148 | 4 | SBDS | AR | p.K62 * | P/LP | HET | P | 120074160 | 45 | 1.67 × 10−4 | 0.996 | - | - | - | 1 | na | BMF | 2 | |
| 209 | M | 144 | 4 | TNFRSF13B | AD/AR | p.C172Y | uncertain significance | HET | LP | 751216929 | 22.2 | 1.90 × 10−4 | 0.7465 | D | D | D | 1 | na | Sanger TERC | ALPS | 1 |
| 220 | M | 141 | 4 | TERT | AD | p.E429 * | - | HET | LP | na | 32 | - | 0.994 | - | - | - | 1 | F | Sanger TERC, TINF2 | BMF | 1 |
| 226 | F | 122 | 6 | TNFRSF13B | AD/AR | p.C104Y | LP | HET | LP | 72553879 | 24.7 | 1.58 × 10−4 | 0.7764 | D | D | D | 2 | na | Neutropenia | 1 | |
| ELANE | AD | p.P139L | P/LP | HET | P | 137854448 | 23.6 | - | 0.999 | D | D | D | 2 | na | 1 | ||||||
| 242 | M | 127 | 2 | TINF2 | AD | p.R282C | P | HET | P | 121918545 | 26.9 | 0.00 | 0.999 | D | D | D | 1 | na | BMF | 1 | |
| 252 | F | 128 | 4 | TNFRSF13B | AD/AR | p.L69T fs*12 | conflicting | HET | LP | 72553875 | 22.8 | 3.09 × 10−4 | - | - | - | - | 3 | na | ALPS | 1 | |
| 253 | M | 154 | 12 | ELANE | AD | p.P139L | P/LP | HET | P | 137854448 | 23.6 | - | 0.999 | D | D | D | 2 | na | Sanger HAX1 | Neutropenia | 1 |
| 1176 | M | 832 | 16 | MVK | AR | p.L168_ D170 delinsHis | uncertain significance | HET | LP | na | - | - | - | - | - | - | 1 | na | Sanger MVK, TNFRSF1A | ALPS-like | 1 |
| p.V377I | conflicting | HET | P ** | 28934897 | 15.11 | 1.47 × 10−3 | 0.981 | D | T | N | 1 | na | |||||||||
| 2130 | F | 1834 | 16 | NOD2 | AD | p.W709 * | - | HET | LP | 776701942 | 36 | 8.03 × 10−6 | 0.985 | - | - | - | 1 | F | PMID: 26386126 | SAID | 1 |
| 260 | F | 118 | 4 | CARD11 | AD | p.M1I | uncertain significance | HET | P | na | 22.4 | - | - | T | D | - | 1 | na | Neutropenia | 1 | |
| 288 | F | 122 | 6 | LRBA | AR | p.Q2717 * | - | HET | P | na | 50 | - | - | - | - | - | 1 | na | ALPS-like | 1 | |
| p.E946 * | - | HET | LP | 777413769 | 24.2 | 3.94 × 10−5 | - | - | - | - | 1 | na | |||||||||
| 285 | M | 132 | 5 | TNFRSF13B | AD/AR | p.I87N | conflicting | HET | LP | 72553877 | 24.6 | 3.48 × 10−4 | - | D | D | - | 1 | na | ALPS-like | 1 | |
| 303 | M | 110 | 4 | FAS | AD | p.H282R fs*14 | - | HET | LP | na | - | - | - | - | - | - | 1 | na | ALPS-like | 1 | |
| 307 | M | 117 | 4 | TNFRSF13B | AD/AR | p.C104R | conflicting | HET | P | 34557412 | 25.8 | 3.92 × 10−3 | 0.9172 | D | D | D | 4 | na | ALPS-like | 1 | |
| 313 | M | 138 | 3 | FAS | AD | p.Gly66C | - | HET | P | na | 34 | - | - | D | D | - | 1 | na | ALPS-like | 1 | |
| 316 | M | 147 | 7 | TNFRSF13B | AD/AR | p.C104R | conflicting | HET | P | 34557412 | 25.8 | 3.92 × 10−3 | 0.9172 | D | D | D | 4 | na | ALPS-like | 1 |
| ID | GENDER | Total Variants Called | Filtered Variants ‡ | Gene | Inherit. of Associated Phenotype | Variant § | CLINVAR | Zygosity | Variant Classific. * | dbSNP (#rs) | CADD Score | Frequency (gnomAD) | DANN Score | FATHMM | SIFT | PROVEAN | Cases with Same Variant | Parental Inherit./de novo | Previous Genetic Tests | Clinical Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | F | 631 | 11 | PRKCD | AR | p.G248S | - | HET | VUS | 144320413 | 28.9 | 6.57 × 10−6 | 0.9989 | D | D | D | 1 | de novo | ALPS-like | |
| 14 | F | 571 | 15 | RAC2 | AD | p.R68Q | - | HET | VUS | na | 29.7 | - | 0.9996 | T | D | D | 1 | na | ALPS-like | |
| 29 | M | 716 | 11 | WRAP53 | AR | p.G481S | - | HET | VUS | 763828661 | 26.6 | 6.58 × 10−6 | 0.9985 | D | D | D | 1 | na | Sanger TERC, TERT | BMF |
| 35 | F | 640 | 13 | LRBA | AR | p.D2294N | - | HET | VUS | 939898061 | 26.3 | 7.97 × 10−6 | 0.999 | T | D | D | 1 | M | Sanger FAS | ALPS-like |
| 40 | F | 534 | 13 | CARD11 | AD | p.R967C | uncertain significance | HET | VUS | 149857605 | 24.8 | 5.26 × 10−5 | 0.9988 | T | D | D | 1 | M | ALPS-like | |
| 48 | M | 594 | 11 | LYST | AR | p.R2624W | conflicting | HET | VUS | 150306354 | 26.3 | 2.81 × 10−3 | 0.9991 | T | D | D | 1 | M | Neutropenia | |
| 71 | F | 1762 | 28 | RAG2 | AR | p.G509D | - | HET | VUS | 779267024 | 15.52 | 7.97 × 10−6 | 0.9969 | D | D | N | 1 | na | Sanger TERC, TERT, TINF2, DKC1 | ALPS-like |
| 73 | F | 996 | 18 | WDR1 | AR | p.T478M | - | HET | VUS | 186889066 | 25.5 | 7.68 × 10−4 | 0.9931 | T | D | D | 1 | na | ALPS-like | |
| 86 | M | 1470 | 21 | ATM | AR | p.R2912G | - | HET | VUS | 376676328 | 26.2 | 2.04 × 10−4 | 0.9986 | D | D | D | 1 | na | BMF | |
| 87 | M | 1827 | 21 | AIRE | AR | p.R356W | - | HET | VUS | 376901046 | 22.3 | 1.45 × 10−4 | 0.9979 | D | D | D | 1 | na | BMF | |
| BLNK | AR | p.G30R | - | HET | VUS | 143109144 | 25.4 | 7.18 × 10−4 | 0.9993 | - | D | D | 1 | na | ||||||
| 88 | F | 1386 | 22 | ATM | AR | p.Y67C | uncertain significance | HET | VUS | 754033733 | 25.6 | 4.02 × 10−6 | 0.9975 | T | D | D | 1 | na | ALPS-like | |
| 90 | M | 1105 | 16 | CXCR4 | AD | p.L125V | - | HET | VUS | 1001278766 | 26.2 | 1.31 × 10−5 | 0.9974 | T | D | D | 1 | na | ALPS-like | |
| 100 | F | 1614 | 22 | TNFRSF13B | AD/AR | p.E117Gfs*35 | uncertain significance | HET | VUS | na | - | - | - | - | - | - | 1 | F | ALPS-like | |
| 102 | M | 1961 | 34 | RAG2 | AR | p.L279P | uncertain significance | HET | VUS | na | 26.7 | - | 0.9985 | D | D | N | 1 | na | ALPS-like | |
| 103 | M | 1509 | 39 | FANCA | AR | p.A430V | uncertain significance | HET | VUS | 772567344 | 22.4 | 6.57 × 10−6 | 0.9947 | D | T | D | 1 | na | Sanger TERC | ALPS-like |
| 110 | F | 1349 | 16 | NLRC4 | AD | p.R492W | uncertain significance | HET | VUS | 1317272776 | 22.3 | 3.98 × 10−6 | 0.9787 | T | D | D | 1 | na | ALPS-like | |
| STAT5B | AD/AR | p.R100C | uncertain significance | HET | VUS | 199894785 | 32 | 7.24 × 10−5 | 0.9994 | T | D | D | 1 | na | ||||||
| 124 | M | 1443 | 19 | CHD7 | AD | p.S1406R | LP | HET | VUS | na | 22.3 | - | 0.995 | T | T | D | 1 | F | BMF | |
| 132 | F | 1607 | 23 | C1S | AD | p.R534W | uncertain significance | HET | VUS | 121909582 | 26.8 | 2.10 × 10−4 | 0.9992 | D | D | D | 1 | na | ALPS-like | |
| 159 | M | 134 | 3 | G6PC | AR | p.T267M | - | HET | VUS | 145296477 | 21.6 | 7.56 × 10−5 | 0.998 | T | T | N | 1 | na | ALPS-like | |
| 172 | M | 133 | 4 | LRBA | AR | p.R2862C | conflicting | HET | VUS | 145709687 | 27.5 | 1.47 × 10−3 | 0.9992 | T | D | D | 1 | na | ALPS-like | |
| 174 | M | 134 | 6 | AP3B1 | AR | p.V315A | uncertain significance | HET | VUS | na | 29.7 | - | 0.9986 | T | D | D | 1 | na | Sanger FAS | ALPS-like |
| 176 | F | 134 | 3 | WAS | XLR | p.E131K | B/LB | HET | VUS | 146220228 | 24.6 | 2.16 × 10−3 | 0.9991 | D | D | D | 1 | na | Sanger FAS | ALPS-like |
| 203 | F | 140 | 6 | TERT | AD/AR | p.E441del | conflicting | HET | VUS | 377639087 | - | 1.72 × 10−3 | - | - | - | - | 1 | na | ALPS | |
| 204 | M | 140 | 6 | ITK | AR | p.Y240C | uncertain significance | HET | VUS | na | 27.4 | - | 0.9982 | T | D | D | 1 | na | AIHA | |
| 205 | M | 155 | 6 | CTC1 | AR | p.P999H | uncertain significance | HET | VUS | 780572571 | 16.72 | 3.19 × 10−5 | 0.9453 | D | D | D | 1 | na | Sanger TERT, TERC | Neutropenia |
| 214 | M | 131 | 4 | CARD11 | AD/AR | p.S439F | uncertain significance | HET | VUS | 760856731 | 28.1 | 2.79 × 10−5 | 0.9979 | T | D | D | 1 | na | Neutropenia | |
| 2130 | F | 1834 | 16 | MPL | AD/AR | p.R537Q | - | HET | VUS | 3820551 | 26 | 9.21 × 10−5 | 0.993 | D | D | N | 1 | na | PMID: 26386126 | SAID |
| 2582 | M | 1703 | 34 | STXBP2 | - | p.I74F | - | HET | VUS | na | 26.6 | - | 0.9899 | D | D | D | 1 | na | PMID: 31325311 | SAID |
| 261 | M | 116 | 2 | CARD11 | AD/AR | p.V90F | - | HET | VUS | na | 25.6 | - | - | D | D | - | 1 | na | ALPS-like | |
| 301 | M | 107 | 9 | PIK3CD | AD/AR | p.P864L | uncertain significance | HET | VUS | 148984508 | 26 | - | - | D | D | - | 1 | na | Neutropenia | |
| 315 | F | 129 | 3 | FAS | AD | p.C135Y | uncertain significance | HET | VUS | na | 25.5 | - | - | D | D | - | 1 | na | ALPS-like |
| A_Variant Distribution among the 272 Patients Studied | |||
| n = 0 | n = 1 | n = 2 | n ≥ 3 |
| 103 | 114 | 38 | 17 |
| 37.9% | 41.9% | 14% | 6.2% |
| B_Classification of the 197 Different Variants Detected | |||
| Pathogenic/Likely Pathogenic | VUS with a probable effect on the phenotype | VUS Low impact/Likely Benign/Benign | |
| 47 | 33 | 117 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossi, A.; Miano, M.; Lanciotti, M.; Fioredda, F.; Guardo, D.; Palmisani, E.; Terranova, P.; Santamaria, G.; Caroli, F.; Caorsi, R.; et al. Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients. Genes 2021, 12, 1299. https://doi.org/10.3390/genes12091299
Grossi A, Miano M, Lanciotti M, Fioredda F, Guardo D, Palmisani E, Terranova P, Santamaria G, Caroli F, Caorsi R, et al. Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients. Genes. 2021; 12(9):1299. https://doi.org/10.3390/genes12091299
Chicago/Turabian StyleGrossi, Alice, Maurizio Miano, Marina Lanciotti, Francesca Fioredda, Daniela Guardo, Elena Palmisani, Paola Terranova, Giuseppe Santamaria, Francesco Caroli, Roberta Caorsi, and et al. 2021. "Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients" Genes 12, no. 9: 1299. https://doi.org/10.3390/genes12091299
APA StyleGrossi, A., Miano, M., Lanciotti, M., Fioredda, F., Guardo, D., Palmisani, E., Terranova, P., Santamaria, G., Caroli, F., Caorsi, R., Volpi, S., Gattorno, M., Dufour, C., & Ceccherini, I. (2021). Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients. Genes, 12(9), 1299. https://doi.org/10.3390/genes12091299