
Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy

Abstract
:1. Introduction
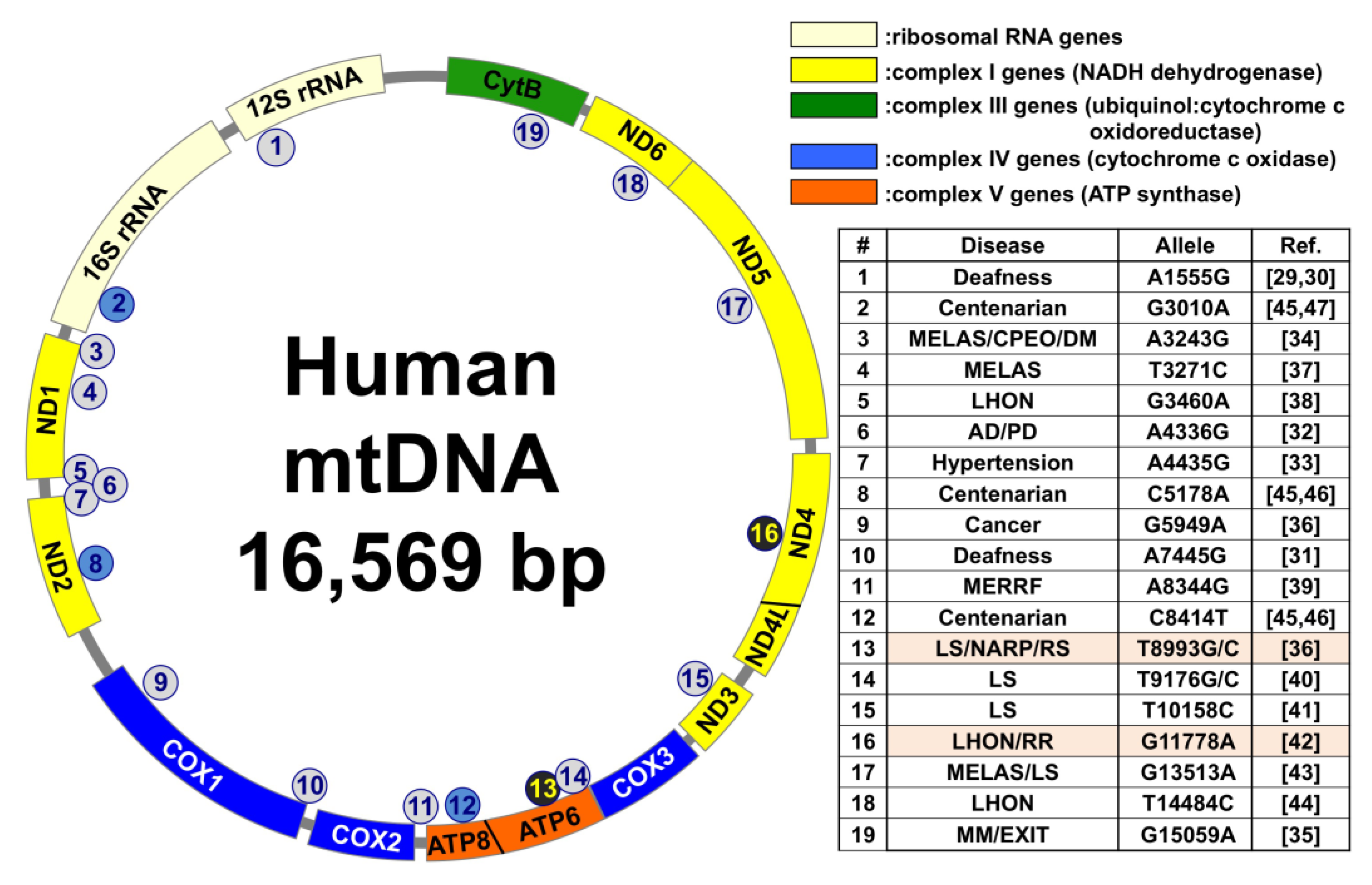
1.1. Association of Mitochondrial DNA (mtDNA) Mutations in Several Diseases, Longevity, and Radioresistance
1.2. mtDNA Copy Number and Its Roles in Disease, Longevity, and Treatment Resistance
1.3. ATP Synthesis, ROS Production, and Mitochondrial Membrane Potential (ΔΨm) in Cancer and Cell Death
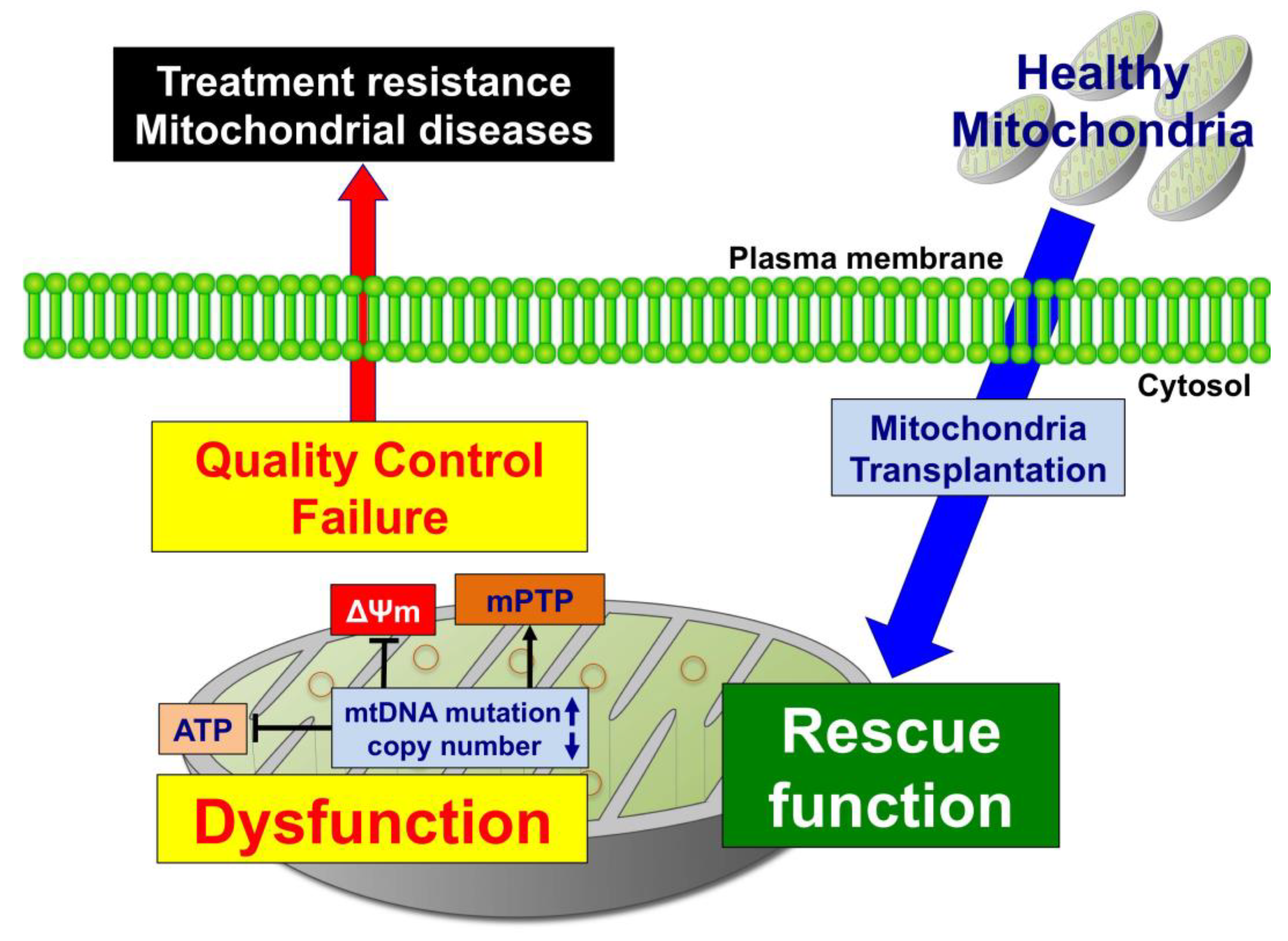
2. Mitochondria Transplantation (mtTP) as a Novel Therapeutic Strategy
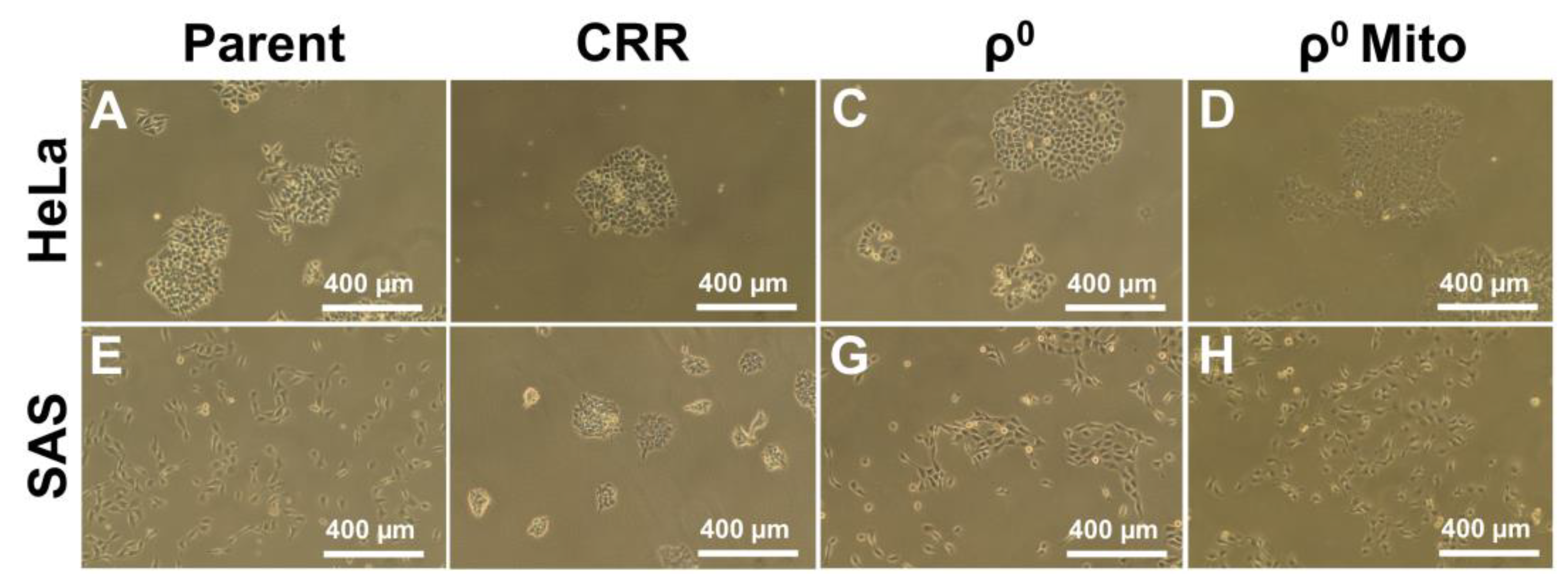
3. Involvement of Mitochondrial Dysfunction in Treatment-Resistant Cancer Cells
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review). Int. J. Oncol. 2003, 22, 15–20. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell. 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Ogura, A.; Oowada, S.; Kon, Y.; Hirayama, A.; Yasui, H.; Meike, S.; Kobayashi, S.; Kuwabara, M.; Inanami, O. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009, 277, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Forkink, M.; Smeitink, J.A.; Brock, R.; Willems, P.H.; Koopman, W.J. Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Biochim. Biophys. Acta 2010, 1797, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K. Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann. N. Y. Acad. Sci. 2004, 1019, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Idelchik, M.D.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Bogenhagen, D.; Clayton, D.A. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 1977, 11, 719–727. [Google Scholar] [CrossRef]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef]
- Wanrooij, S.; Falkenberg, M. The human mitochondrial replication fork in health and disease. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1378–1388. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000, 15 (Suppl. 2), 11–17. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Ann. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Shimoizumi, H.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. Respiration-deficient cells are caused by a single point mutation in the mitochondrial tRNA-Leu (UUR) gene in mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS). Am. J. Hum. Genet. 1991, 49, 590–599. [Google Scholar]
- Lorenzoni, P.J.; Werneck, L.C.; Kay, C.S.; Silvado, C.E.; Scola, R.H. When should MELAS (Mitochondrial myopathy, Encephalopathy, Lactic Aci- dosis, and Stroke-like episodes) be the diagnosis? Arq. Neuro-Psiquiatr. 2015, 73, 959–967. [Google Scholar] [CrossRef]
- Dimauro, S. Mitochondrial diseases. Biochim. Biophys. Acta 2004, 1658, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.; Silvado, C.E.; Werneck, L.C. When should MERRF (myoclonus epilepsy associated with ragged-red fibers) be the diagnosis? Arq. Neuro-Psiquiatr. 2014, 72, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabunga, P.; Lau, A.K.; Phan, K.; Puranik, R.; Liang, C.; Davis, R.L.; Sue, C.M.; Sy, R.W. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int. J. Cardiol. 2015, 181, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Ciacci, F.; Silvestri, G.; Shanske, S.; Sciacco, M.; Hirano, M.; Schon, E.A.; Bonilla, E.; DiMauro, S. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul. Disord. 1993, 3, 43–50. [Google Scholar] [CrossRef]
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns–Sayre syndrome. N. Engl. J. Med. 1989, 320, 1293–1299. [Google Scholar] [CrossRef]
- Van Goethem, G.; Martin, J.J.; van Broeckhoven, C. Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: Unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromol. Med. 2003, 3, 129–146. [Google Scholar] [CrossRef]
- Morris, A.A.; Leonard, J.V.; Brown, G.K.; Bidouki, S.K.; Bindoff, L.A.; Woodward, C.E.; Harding, A.E.; Lake, B.D.; Harding, B.N.; Farrell, M.A.; et al. Deficiency of respiratory chain complex I is a common cause of Leigh disease. Ann. Neurol. 1996, 40, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Van den Ouweland, J.M.; Lemkes, H.H.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.; van de Kamp, J.J.; Maassen, J.A. Mutation in mitochondrial tRNA (Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar] [CrossRef]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Oztas, S.; Qiu, W.Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Reid, F.M.; Vernham, G.A.; Jacobs, H.T. A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum. Mutat. 1994, 3, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Otaegui, D.; Paisán, C.; Sáenz, A.; Martí, I.; Ribate, M.; Martí-Massó, J.F.; Pérez-Tur, J.; López de Munain, A. Mitochondrial polymporphisms in Parkinson’s Disease. Neurosci. Lett. 2004, 370, 171–174. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Li, Z.; Wang, X.J.; Yang, L.; Wang, S.; Guan, M.X. Mitochondrial transfer RNAMet 4435A>G mutation is associated with maternally inherited hypertension in a Chinese pedigree. Hypertension 2009, 53, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef]
- Rose, M.R. Mitochondrial myopathies: Genetic mechanisms. Arch. Neurol. 1998, 55, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, M.A.; Maguire, J.; Myint, T.; Applegarth, D.; Robinson, B.H. Clinical, physiological, and histological features in a kindred with the T3271C melas mutation. Muscle Nerve 1998, 21, 25–33. [Google Scholar] [CrossRef]
- Macmillan, C.; Kirkham, T.; Fu, K.; Allison, V.; Andermann, E.; Chitayat, D.; Fortier, D.; Gans, M.; Hare, H.; Quercia, N.; et al. Pedigree analysis of French Canadian families with T14484C Leber’s hereditary optic neuropathy. Neurology 1998, 50, 417–422. [Google Scholar] [CrossRef]
- Larsson, N.G.; Tulinius, M.H.; Holme, E.; Oldfors, A. Pathogenetic aspects of the A8344G mutation of mitochondrial DNA associated with MERRF syndrome and multiple symmetric lipomas. Muscle Nerve Suppl. 1995, 3, S102–S106. [Google Scholar] [CrossRef]
- Carrozzo, R.; Tessa, A.; Vázquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schägger, H.; et al. The T9176G mtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology 2001, 56, 687–690. [Google Scholar] [CrossRef]
- Crimi, M.; Papadimitriou, A.; Galbiati, S.; Palamidou, P.; Fortunato, F.; Bordoni, A.; Papandreou, U.; Papadimitriou, D.; Hadjigeorgiou, G.M.; Drogari, E.; et al. A new mitochondrial DNA mutation in ND3 gene causing severe Leigh syndrome with early lethality. Pediatr. Res. 2004, 55, 842–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.; De Coo, I.F.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Yahata, N.; Matsumoto, Y.; Omi, M.; Yamamoto, N.; Hata, R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep. 2017, 7, 15557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, N.; Herrnstadt, C.; Shults, C.; Mackey, D.A. Low penetrance of the 14484 LHON mutation when it arises in a non-haplogroup J mtDNA background. Am. J. Med. Genet A 2003, 119A, 147–151. [Google Scholar] [CrossRef]
- Tanaka, M.; Gong, J.S.; Zhang, J.; Yoneda, M.; Yagi, K. Mitochondrial genotype associated with longevity. Lancet 1998, 351, 185–186. [Google Scholar] [CrossRef]
- Gong, J.-S.; Zhang, J.; Yoneda, M.; Sahashi, K.; Miyajima, H.; Yamauchi, K.; Yagi, K.; Tanaka, M. Mitochondrial Genotype Frequent in Centenarians Predisposes Resistance to Adult-Onset Diseases. J. Clin. Biochem. Nutr. 1998, 24, 105–111. [Google Scholar] [CrossRef]
- Kang, D.; Takashige, K.; Sekiguchi, M.; Singh, K.K. Mitochondrial DNA Mutations in aging Disease and Cancer; Springer Nature Switzerland AG: Cham, Switzerland, 1998; Chapter 1; pp. 1–15. [Google Scholar]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Ann. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Takagi, K.; Yamada, Y.; Gong, J.S.; Sone, T.; Yokota, M.; Tanaka, M. Association of a 5178C-->A (Leu237Met) polymorphism in the mitochondrial DNA with a low prevalence of myocardial infarction in Japanese individuals. Atherosclerosis 2004, 175, 281–286. [Google Scholar] [CrossRef]
- Simonetti, S.; Chen, X.; DiMauro, S.; Schon, E.A. Accumulation of deletions in human mitochondrial DNA during normal aging: Analysis by quantitative PCR. Biochim. Biophys. Acta. 1992, 1180, 113–122. [Google Scholar] [CrossRef]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Kulkarni, R.; Reither, A.; Thomas, R.A.; Tucker, J.D. Mitochondrial mutant cells are hypersensitive to ionizing radiation, phleomycin and mitomycin C. Mutat. Res. 2009, 663, 46–51. [Google Scholar] [CrossRef]
- Mengel-From, J.; Thinggaard, M.; Dalgárd, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knez, J.; Winckelmans, E.; Plusquin, M.; Thijs, L.; Cauwenberghs, N.; Gu, Y.; Staessen, J.A.; Nawrot, T.S.; Kuznetsova, T. Correlates of peripheral blood mitochondrial DNA content in a general population. Am. J. Epidemiol. 2016, 183, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Sondheimer, N.; Glatz, C.E.; Tirone, J.E.; Deardorff, M.A.; Krieger, A.M.; Hakonarson, H. Neutral mitochondrial heteroplasmy and the influence of aging. Hum. Mol. Genet. 2011, 20, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Rothwell, R.; Vermaat, M.; Wachsmuth, M.; Schroder, R.; Laros, J.F.; van Oven, M.; De Bakker, P.I.; Bovenberg, J.A.; van Duijn, C.M.; et al. Transmission of human mtDNA heteroplasmy in the genome of the Netherlands families: Support for a variable-size bottleneck. Genome Res. 2016, 26, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indo, H.P.; Suenaga, S.; Tomita, K.; Ito, H.; Matsui, H.; Majima, H.J. Analysis of Oxidative Stress Marker, mitochondrial DNA copy numbers and Mitochondrial DNA Oxidation among 135 persons who live in Amami islands, high centenarian population District in Kagoshima. Free Radic. Biol. Med. 2018, 120, S134. [Google Scholar] [CrossRef]
- Sun, X.; Zhan, L.; Chen, Y.; Wang, G.; He, L.; Wang, Q.; Zhou, F.; Yang, F.; Wu, J.; Wu, Y.; et al. Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal. Transduct. Target. Ther. 2018, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Mi, J.; Tian, G.; Liu, S.; Li, X.; Ni, T.; Zhang, L.; Wang, B. The relationship between altered mitochondrial DNA copy number and cancer risk: A meta-analysis. Sci. Rep. 2015, 5, 10039. [Google Scholar] [CrossRef] [Green Version]
- Lemnrau, A.; Brook, M.N.; Fletcher, O.; Coulson, P.; Tomczyk, K.; Jones, M.; Ashworth, A.; Swerdlow, A.; Orr, N.; Garcia-Closas, M. Mitochondrial DNA copy number in peripheral blood cells and risk of developing breast cancer. Cancer Res. 2015, 75, 2844–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Wan, J.; Song, R.; Zhao, H. Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: A replication study. Carcinogenesis 2015, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Gopalakrishnan, V.; Lee, J.E.; Fang, S.; Zhao, H. Mitochondrial DNA copy number in peripheral blood and melanoma risk. PLoS ONE 2015, 10, e0131649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosgood, H.D., 3rd; Liu, C.-S.; Rothman, N.; Weinstein, S.J.; Bonner, M.R.; Shen, M.; Lim, U.; Virtamo, J.; Cheng, W.; Albanes, D.; et al. Mitochondrial DNA copy number and lung cancer risk in a prospective cohort study. Carcinogenesis 2010, 31, 847–849. [Google Scholar] [CrossRef]
- Lynch, S.M.; Weinstein, S.J.; Virtamo, J.; Lan, Q.; Liu, C.-S.; Cheng, W.-L.; Rothman, N.; Albanes, D.; Stolzenberg- Solomon, R.Z. Mitochondrial DNA copy number and pancreatic cancer in the Alpha-tocopherol beta- carotene cancer prevention study. Cancer Prev. Res. 2011, 4, 1912–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Chen, M.; Wood, C.G.; Lin, J.; Spitz, M.R.; Ma, J.; Amos, C.I.; Shields, P.G.; Benowitz, N.L.; Gu, J.; et al. Mitochondrial DNA content: Its genetic heritability and association with renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Huang, B.; Gao, Y.-T.; Shu, X.-O.; Wen, W.; Yang, G.; Li, G.; Courtney, R.; Ji, B.-T.; Li, H.-L.; Purdue, M.P.; et al. Association of leukocyte mitochondrial DNA copy number with colorectal cancer risk: Results from the Shanghai Women’s Health Study. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2357–2365. [Google Scholar] [CrossRef] [Green Version]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Park, S.Y.; Chang, I.; Kim, J.Y.; Kang, S.W.; Park, S.H.; Singh, K.; Lee, M.S. Resistance of mitochondrial DNA-depleted cells against cell death: Role of mitochondrial superoxide dismutase. J. Biol. Chem. 2004, 279, 7512–7520. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Tsuji, M.; Kawamoto, T.; Yamazaki, T. Involvement of reactive ox- ygen species derived from mitochondria in neuronal injury elicited by methylmercury. J. Clin. Biochem. Nutr. 2016, 59, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Mitochondrial function is required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. FEBS Lett. 1997, 410, 219–222. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Rego, A.C.; Penacho, N.; Oliveira, C.R. Apoptotic cell death induced by hydrogen peroxide in NT2 parental and mitochondrial DNA depleted cells. Neurochem. Int. 2004, 45, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Kuwahara, Y.; Takashi, Y.; Tsukahara, T.; Kurimasa, A.; Fukumoto, M.; Nishitani, Y.; Sato, T. Sensitivity of mitochondrial DNA depleted ρ0 cells to H2O2 depends on the plasma membrane status. Biochem. Biophys. Res. Commun. 2017, 490, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Minakami, S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J. 1979, 180, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Takayanagi, R.; Takeshige, K.; Minakami, S. NADH- and NADPH-dependent lipid peroxidation in bovine heart submitochondrial particles. Dependence on the rate of electron flow in the respiratory chain and an antioxidant role of ubiquinol. Biochem. J. 1980, 192, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Zanin, C.; Vayssière, J.L.; Petit, P.X.; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995, 181, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondrial organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, H.; Roushandeh, A.M.; Rostami, M.K.; Razavi-Toosi, M.T.; Shokrgozar, M.A.; Jahanian-Najafabadi, A.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat. Biochim. Biophys. Acta (BBA) 2020, 1866, 165809. [Google Scholar] [CrossRef]
- Roushandeh, A.M.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial transplantation as a potential and novel master key for treatment of various incurable diseases. Cytotechnology 2019, 71, 647–663. [Google Scholar] [CrossRef]
- Pourmohammadi-Bejarpasi, Z.; Roushandeh, A.M.; Saberi, A.; Kheirandish-Rostami, M.; Toosi, S.M.R.; Jahanian-Najafabadi, A.; Tomita, K.; Kuwahara, Y.; Sato, T.; Roudkenar, M.H. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res. Bull. 2020, 165, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Roushandeh, A.M.; Tomita, K.; Kuwahara, Y.; Jahanian-Najafabadi, A.; Igarashi, K.; Roudkenar, M.H.; Sato, T. Transfer of healthy fibroblast-derived mitochondria to HeLa ρ0 and SAS ρ0 cells recovers the proliferation capabilities of these cancer cells under conventional culture medium but increase their sensitivity to cisplatin-induced apoptotic death. Mol. Biol. Rep. 2020, 47, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Kheirandish-Rostami, M.; Roudkenar, M.H.; Jahanian-Najafabadi, A.; Tomita, K.; Kuwahara, Y.; Sato, T.; Roushandeh, A.M. Mitochondrial characteristics contribute to proliferation and migration potency of MDA-MB-231 cancer cells and their response to cisplatin treatment. Life Sci. 2020, 244, 117339. [Google Scholar] [CrossRef] [PubMed]
- Takashi, Y.; Tomita, K.; Kuwahara, Y.; Roudkenar, M.H.; Roushandeh, A.M.; Igarashi, K.; Nagasawa, T.; Nishitani, Y.; Sato, T. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free Radic. Biol. Med. 2020, 161, 60–70. [Google Scholar] [CrossRef]
- Guariento, A.; Doulamis, I.P.; Duignan, T.; Kido, T.; Regan, W.L.; Saeed, M.Y.; Hoganson, D.M.; Emani, S.M.; Fynn-Thompson, F.; Matte, G.S.; et al. Mitochondrial transplantation for myocardial protection in ex-situ-perfused hearts donated after circulatory death. J. Heart Lung Transplant. 2020, 2498, 31625–31629. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Callaway, E. UK sets sights on gene therapy in eggs. Nature 2012, 481, 419. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.H.; Liu, Z.; Li, H.; Zhu, L.; Chen, S.L.; Xing, F.Q. First twins born in Mainland China by autologous granular cell mitochondria transfer. Acad. J. First Med. Coll. PLA 2003, 23, 990–991. [Google Scholar]
- Shin, B.; Cowan, D.B.; Emani, S.M.; Del Nido, P.J.; McCully, J.D. Mitochondrial Transplantation in Myocardial Ischemia and Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 595–619. [Google Scholar]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Kuo, S.J.; Chen, S.T.; Liu, C.S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Wu, S.L.; Liu, K.H.; Chen, Y.H.; Chuang, C.S.; Cheng, F.C.; Su, H.L.; Wei, Y.H.; Kuo, S.J.; Liu, C.S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 2016, 170, 40–56. [Google Scholar] [CrossRef]
- Kuo, C.C.; Su, H.L.; Chang, T.L.; Chiang, C.Y.; Sheu, M.L.; Cheng, F.C.; Chen, C.J.; Sheehan, J.; Pan, H.C. Prevention of Axonal Degeneration by Perineurium Injection of Mitochondria in a Sciatic Nerve Crush Injury Model. Neurosurgery 2017, 80, 475–488. [Google Scholar] [CrossRef]
- Woods, D.C.; Tilly, J.L. Autologous Germline Mitochondrial Energy Transfer (AUGMENT) in human assisted reproduction. Semin. Reprod. Med. 2015, 33, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Qin, A.; Liu, D.; Ruan, R.; Wang, Q.; Yuan, J.; Cheng, T.S.; Filipovska, A.; Papadimitriou, J.M.; Dai, K.; et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 2019, 5, eaaw7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, Y.; Li, L.; Baba, T.; Nakagawa, H.; Shimura, T.; Yamamoto, Y.; Ohkubo, Y.; Fukumoto, M. Clinically relevant radioresistant cells efficiently repair DNA double-strand breaks induced by X-rays. Cancer Sci. 2009, 100, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Mori, M.; Oikawa, T.; Shimura, T.; Ohtake, Y.; Mori, S.; Ohkubo, Y.; Fukumoto, M. The modified high-density survival assay is the useful tool to predict the effectiveness of fractionated radiation exposure. J. Radiat. Res. 2010, 51, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, Y.; Mori, M.; Kitahara, S.; Fukumoto, M.; Ezaki, T.; Mori, S.; Echigo, S.; Ohkubo, Y.; Fukumoto, M. Targeting of tumor endothelial cells combining 2 Gy/day of X-ray with Everolimus is the effective modality for overcoming clinically relevant radioresistant tumors. Cancer Med. 2014, 3, 310–321. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Roudkenar, M.H.; Urushihara, Y.; Saito, Y.; Tomita, K.; Roushandeh, A.M.; Sato, T.; Kurimasa, A.; Fukumoto, M. Clinically relevant radioresistant cells: A simple model to understand cancer radioresistance. Med. Mol. Morphol. 2017, 50, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Kuwahara, Y.; Takashi, Y.; Igarashi, K.; Nagasawa, T.; Nabika, H.; Kurimasa, A.; Fukumoto, M.; Nishitani, Y.; Sato, T. Clinically Relevant Radioresistant Cells Exhibit Resistance to H2O2 by Decreasing Internal H2O2 and Lipid Peroxidation. Tumor Biol. 2018, 40, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallast, S.; Arai, K.; Wang, X.; Lo, E.H.; van Leyen, K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J. Neurochem. 2009, 111, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Nagasawa, T.; Kuwahara, Y.; Torii, S.; Igarashi, K.; Roudkenar, M.H.; Roushandeh, A.M.; Kurimasa, A.; Sato, T. MiR-7-5p Is Involved in Ferroptosis Signaling and Radioresistance Thru the Generation of ROS in Radioresistant HeLa and SAS Cell Lines. Int. J. Mol. Sci. 2021, 22, 8300. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Tomita, K.; Roudkenar, M.H.; Roushandeh, A.M.; Urushihara, Y.; Igarashi, K.; Nagasawa, T.; Kurimasa, A.; Fukumoto, M.; Sato, T. The Effects of Hydrogen Peroxide and/or Radiation on the Survival of Clinically Relevant Radioresistant Cells. Technol Cancer Res. Treat. 2020, 19, 1533033820980077. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Roudkenar, M.H.; Suzuki, M.; Urushihara, Y.; Fukumoto, M.; Saito, Y.; Fukumoto, M. The involvement of mitochondrial membrane potential in cross-resistance between radiation and docetaxel. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 556–565. [Google Scholar] [CrossRef]
- Tomita, K.; Fukumoto, M.; Itoh, K.; Kuwahara, Y.; Igarashi, K.; Nagasawa, T.; Suzuki, M.; Kurimasa, A.; Sato, T. MiR-7-5p is a key factor that controls radioresistance via intracellular Fe2+ content in clinically relevant radioresistant cells. Biochem. Biophys. Res. Commun. 2019, 518, 712–718. [Google Scholar] [CrossRef]
- Fukumoto, M.; Amanuma, T.; Kuwahara, Y.; Shimura, T.; Suzuki, M.; Mori, S.; Kumamoto, H.; Saito, Y.; Ohkubo, Y.; Duan, Z.; et al. Guanine nucleotide-binding protein 1 is one of the key molecules contributing to cancer cell radioresistance. Cancer Sci. 2014, 105, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Guo, H.; Yang, J.; Ji, Y.; Wu, C.S.; Chen, X. Down-regulation of guanylate binding protein 1 causes mitochondrial dysfunction and cellular senescence in macrophages. Sci. Rep. 2018, 8, 1679. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Oikawa, T.; Ochiai, Y.; Roudkenar, M.H.; Fukumoto, M.; Shimura, T.; Ohtake, Y.; Ohkubo, Y.; Mori, S.; Uchiyama, Y.; et al. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2011, 2, e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Takashi, Y.; Tomita, K.; Kuwahara, Y.; Nabika, H.; Igarashi, K.; Nagasawa, T.; Kurimasa, A.; Fukumoto, M.; Nishitani, Y.; Sato, T. Data on the aquaporin gene expression differences among ρ0, clinically relevant radioresistant, and the parental cells of human cervical cancer and human tongue squamous cell carcinoma. Data Brief. 2018, 20, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Petragnano, F.; Pietrantoni, I.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; Macioce, G.; Giordani, I.; Tini, P.; Cheleschi, S.; et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: Functional, molecular and immune-related characterization. J. Biomed. Sci. 2020, 27, 90. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Donor | Recipient | Disease | Result | Reference |
|---|---|---|---|---|
| Rectus Abdominis | Heart | Heart ischemia reperfusion | Cardiac function improved | [85,98] |
| Granular cells | Oocyte | Infertility | Normal babies were born | [101] |
| Astrocytes | Neuron | Ischemic damage | Recover ATP production | [104] |
| HeLa cells (cervical cancer cell line) | AD model mice | Alzheimer disease | Cognitive defect and gliosis were ameliorated | [105] |
| Cybrids from PC-12 cells and human osteosarcoma | Brain | 6-OHDA induced PD model | Improve motor function and mitochondrial function | [106] |
| BHK-21 cell (kidney cell line) | Sciatic nerve | Sciatic nerve crush | Injured sciatic nerve improved | [107] |
| Oocyte cytoplasm | Oocyte | Infertility | Increase pregnancy | [108] |
| Mesenchymal stem cells | Brain | Rat brain ischemia reperfusion | Protect from apoptosis Restores motor function | [94] |
| WI-38 (fibroblast cell line) | ρ0 cells (HeLa, SAS) | mtDNA deficient | Prohibitin 2 enhancement Survive without pyruvate and uridine | [97] |
| MLO-Y4 cell (osteocyte cell line) | ρ0 cells (MLO-Y4) | mtDNA deficient | Increase ATP production | [109] |
| CRR Characteristics | References | |
|---|---|---|
| Morphology | Tight binding | This review, [116] |
| Irradiation | Resistant | [111,117] |
| H2O2 | Resistant | [114] |
| Docetaxel | Resistant | [118] |
| DNA DSB | Low | [112] |
| ΔΨm | Low | [118] |
| Superoxide | Low | [114] |
| Hydroxyl radical | Low | [114] |
| Lipid peroxidation | Low | [114] |
| mtDNA copy number | Low | [114] |
| ATP production | Low | [114] |
| Fe2+ amount | Low | [119] |
| AQP8 gene expression | Low | [114] |
| ALOX gene expression | Low | [114] |
| GBP1 gene expression | High | [120] |
| miR-7-5p expression | High | [119] |
| Localization | Gene Name | ||
|---|---|---|---|
| Plasma membrane | ATP2B2 | FLRT2 | SEMA4C |
| SEAMA6D | TMEM65 | VSTM4 | |
| Cytoplasm | AKT3 | MAPK4 | - |
| Mitochondria | CRLS1 | NDFUA4 | PTPMT1 |
| SLC25A15 | SLC25A16 | SLC25A37 | |
| TIMM50 | TMEM65 | VDAC1 | |
| ER | SERP1 | - | - |
| Lysosome | BLOC1S4 | - | - |
| Golgi apparatus | GLG1 | GOLGB1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomita, K.; Kuwahara, Y.; Igarashi, K.; Roudkenar, M.H.; Roushandeh, A.M.; Kurimasa, A.; Sato, T. Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy. Genes 2021, 12, 1348. https://doi.org/10.3390/genes12091348
Tomita K, Kuwahara Y, Igarashi K, Roudkenar MH, Roushandeh AM, Kurimasa A, Sato T. Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy. Genes. 2021; 12(9):1348. https://doi.org/10.3390/genes12091348
Chicago/Turabian StyleTomita, Kazuo, Yoshikazu Kuwahara, Kento Igarashi, Mehryar Habibi Roudkenar, Amaneh Mohammadi Roushandeh, Akihiro Kurimasa, and Tomoaki Sato. 2021. "Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy" Genes 12, no. 9: 1348. https://doi.org/10.3390/genes12091348
APA StyleTomita, K., Kuwahara, Y., Igarashi, K., Roudkenar, M. H., Roushandeh, A. M., Kurimasa, A., & Sato, T. (2021). Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy. Genes, 12(9), 1348. https://doi.org/10.3390/genes12091348