
Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications


{kind=link}
{kind=link}
Abstract
:1. Introduction
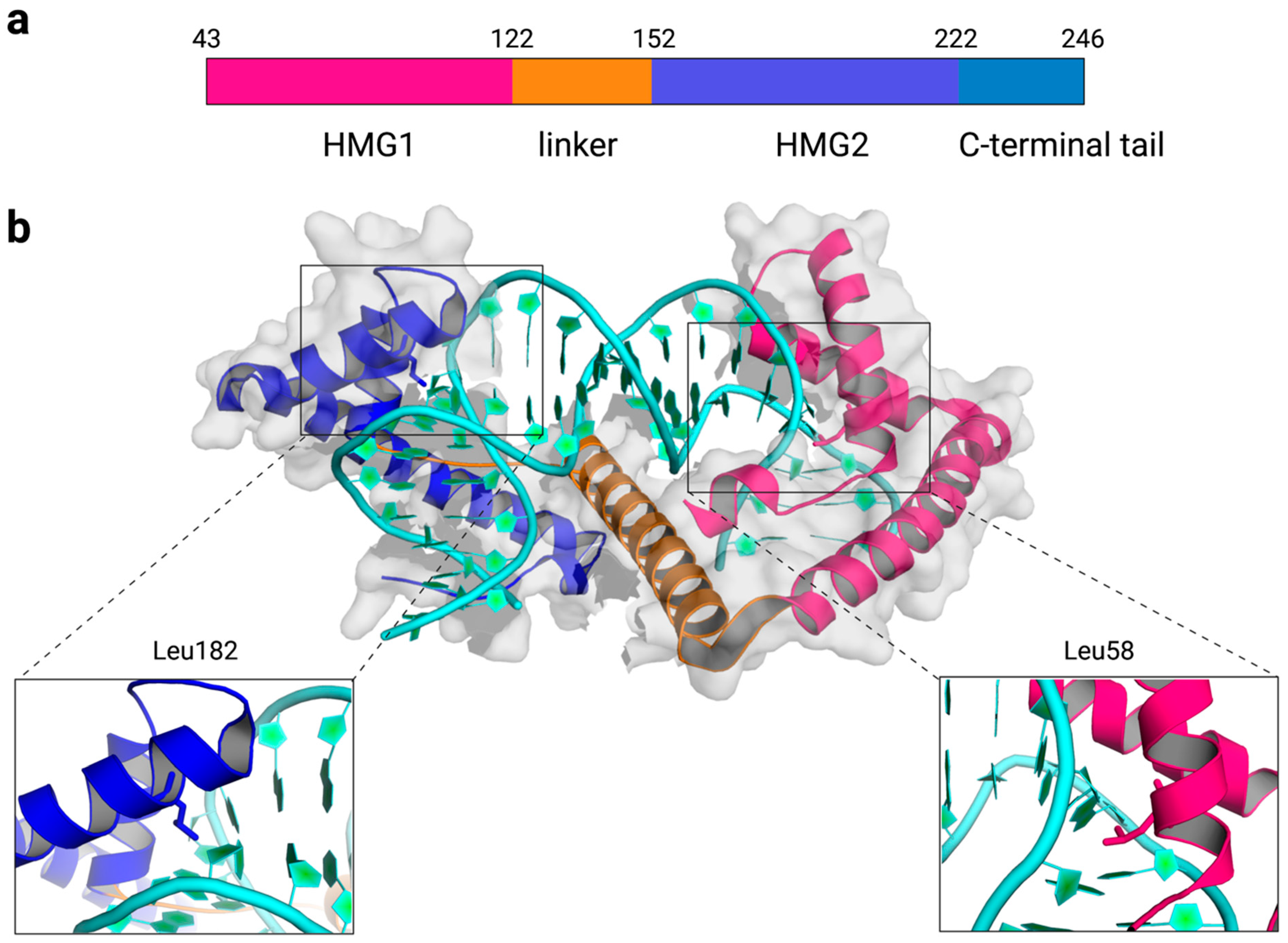
2. Structures of TFAM-DNA Complexes
3. Dynamics of TFAM-DNA Interactions
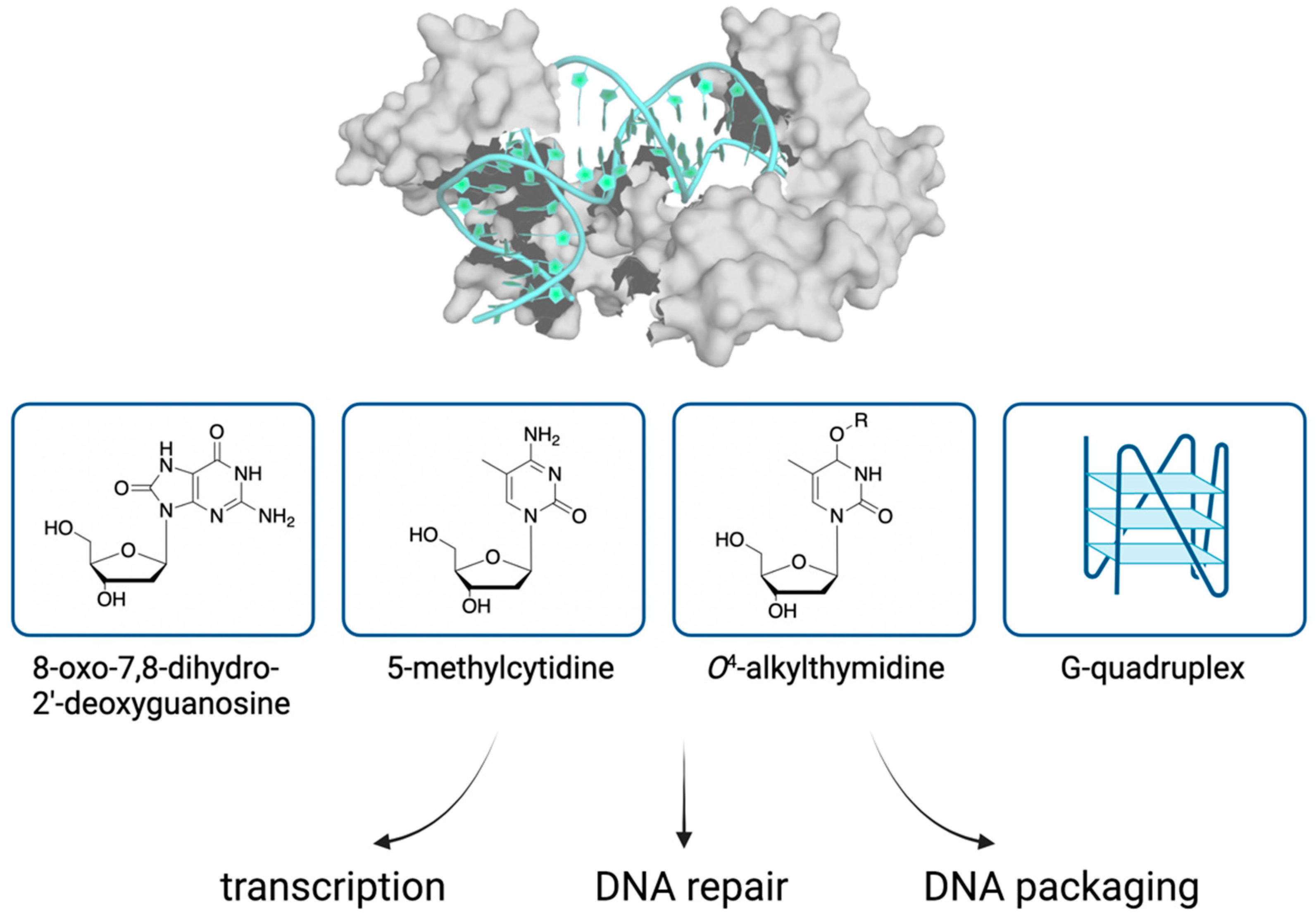
4. Binding of TFAM to Non-Canonical DNA Structures
4.1. Oxidative Damage
4.2. DNA Methylation
4.3. O4-Alkylthymidine DNA Lesions
4.4. Abasic Sites
4.5. Other Non-Canonical DNA Structures
5. Post-Translation Modifications of TFAM
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Sumberaz, P. Mitochondrial DNA Damage: Prevalence, Biological Consequence, and Emerging Pathways. Chem. Res. Toxicol. 2020, 33, 2491–2502. [Google Scholar] [CrossRef]
- Zhao, L. Chapter Ten—Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. In The Enzymes; Zhao, L., Kaguni, L.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 45, pp. 311–341. [Google Scholar]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Doublié, S. Base excision repair in the mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Alencar, R.R.; Batalha, C.; Freire, T.S.; de Souza-Pinto, N.C. Enzymology of mitochondrial DNA repair. Enzymes 2019, 45, 257–287. [Google Scholar]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spåhr, H.; Bonekamp, N.A.; Kühl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Hensen, F.; Cansiz, S.; Gerhold, J.M.; Spelbrink, J.N. To be or not to be a nucleoid protein: A comparison of mass-spectrometry based approaches in the identification of potential mtDNA-nucleoid associated proteins. Biochimie 2014, 100, 219–226. [Google Scholar] [CrossRef]
- Han, S.; Udeshi, N.D.; Deerinck, T.J.; Svinkina, T.; Ellisman, M.H.; Carr, S.A.; Ting, A.Y. Proximity biotinylation as a method for mapping proteins associated with mtDNA in living cells. Cell Chem. Biol. 2017, 24, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, C.; Umeda, S.; Ohsato, T.; Ohno, T.; Abe, Y.; Fukuoh, A.; Shinagawa, H.; Hamasaki, N.; Kang, D. Regulation of mitochondrial D-loops by transcription factor A and single-stranded DNA-binding protein. EMBO Rep. 2002, 3, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Canugovi, C.; Maynard, S.; Bayne, A.-C.V.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Rubio-Cosials, A.; Solà, M. U-turn DNA bending by human mitochondrial transcription factor A. Curr. Opin. Struct. Biol. 2013, 23, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM–mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [Green Version]
- Vozáriková, V.; Kunová, N.; Bauer, J.A.; Frankovský, J.; Kotrasová, V.; Procházková, K.; Džugasová, V.; Kutejová, E.; Pevala, V.; Nosek, J. Mitochondrial HMG-Box Containing Proteins: From Biochemical Properties to the Roles in Human Diseases. Biomolecules 2020, 10, 1193. [Google Scholar] [CrossRef]
- Ngo, H.B.; Kaiser, J.T.; Chan, D.C. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat. Struct. Mol. Biol. 2011, 18, 1290–1296. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Cosials, A.; Sydow, J.F.; Jiménez-Menéndez, N.; Fernández-Millán, P.; Montoya, J.; Jacobs, H.T.; Coll, M.; Bernadó, P.; Solà, M. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nat. Struct. Mol. Biol. 2011, 18, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [Green Version]
- Cuppari, A.; Fernández-Millán, P.; Battistini, F.; Tarrés-Solé, A.; Lyonnais, S.; Iruela, G.; Ruiz-Lopez, E.; Enciso, Y.; Rubio-Cosials, A.; Prohens, R. DNA specificities modulate the binding of human transcription factor A to mitochondrial DNA control region. Nucleic Acids Res. 2019, 47, 6519–6537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Cosials, A.; Battistini, F.; Gansen, A.; Cuppari, A.; Bernadó, P.; Orozco, M.; Langowski, J.; Tóth, K.; Solà, M. Protein flexibility and synergy of HMG domains underlie U-turn bending of DNA by TFAM in solution. Biophys. J. 2018, 114, 2386–2396. [Google Scholar] [CrossRef] [Green Version]
- Farge, G.; Laurens, N.; Broekmans, O.D.; Van Den Wildenberg, S.M.; Dekker, L.C.M.; Gaspari, M.; Gustafsson, C.M.; Peterman, E.J.G.; Falkenberg, M.; Wuite, G.J.L. Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat. Commun. 2012, 3, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, I.; Sitters, G.; Broekmans, O.D.; Farge, G.; Menges, C.; Wende, W.; Hell, S.W.; Peterman, E.J.; Wuite, G.J. STED nanoscopy combined with optical tweezers reveals protein dynamics on densely covered DNA. Nat. Methods 2013, 10, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feric, M.; Demarest, T.G.; Tian, J.; Croteau, D.L.; Bohr, V.A.; Misteli, T. Self-assembly of multi-component mitochondrial nucleoids via phase separation. EMBO J. 2021, 40, e107165. [Google Scholar] [CrossRef]
- Blumberg, A.; Danko, C.G.; Kundaje, A.; Mishmar, D. A common pattern of DNase I footprinting throughout the human mtDNA unveils clues for a chromatin-like organization. Genome Res. 2018, 28, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Murugesapillai, D.; McCauley, M.J.; Maher, L.J.; Williams, M.C. Single-molecule studies of high-mobility group B architectural DNA bending proteins. Biophys. Rev. 2016, 9, 17–40. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.S.; Rajagopalan, S.; Freund, S.M.; Rutherford, T.J.; Andreeva, A.; Townsley, F.M.; Petrovich, M.; Fersht, A.R. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009, 37, 6765–6783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traverso, J.J.; Manoranjan, V.S.; Bishop, A.R.; Rasmussen, K.Ø.; Voulgarakis, N.K. Allostery through protein-induced DNA bubbles. Sci. Rep. 2015, 5, 9037. [Google Scholar] [CrossRef] [Green Version]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.N.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 2011, 120, S130–S145. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- de Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar] [PubMed]
- Kakimoto, M.; Inoguchi, T.; Sonta, T.; Yu, H.Y.; Imamura, M.; Etoh, T.; Hashimoto, T.; Nawata, H. Accumulation of 8-hydroxy-2′-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 2002, 51, 1588–1595. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, H.; Kaneko, T.; Tahara, S.; Hayashi, E.; Naito, H.; Radak, Z.; Goto, S. Regular exercise reduces 8-oxodG in the nuclear and mitochondrial DNA and modulates the DNA repair activity in the liver of old rats. Exp. Gerontol. 2007, 42, 287–295. [Google Scholar] [CrossRef]
- Suter, M.; Richter, C. Fragmented mitochondrial DNA is the predominant carrier of oxidized DNA bases. Biochemistry 1999, 38, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; Collins, L.B.; Liao, S.; Luke, A.M.; Pachkowski, B.F.; Watters, J.L.; Swenberg, J.A. Analysis of 8-oxo-7, 8-dihydro-2′-deoxyguanosine by ultra high pressure liquid chromatography–heat assisted electrospray ionization–tandem mass spectrometry. J. Chromatogr. B 2010, 878, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Izumi, H.; Ise, T.; Uramoto, H.; Torigoe, T.; Ishiguchi, H.; Murakami, T.; Tanabe, M.; Nakayama, Y.; Itoh, H. Human mitochondrial transcription factor A binds preferentially to oxidatively damaged DNA. Biochem. Biophys. Res. Commun. 2002, 295, 945–951. [Google Scholar] [CrossRef]
- Maresca, A.; Zaffagnini, M.; Caporali, L.; Carelli, V.; Zanna, C. DNA methyltransferase 1 mutations and mitochondrial pathology: Is mtDNA methylated? Front Genet. 2015, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.-M.; Baccarelli, A.A. Environmental exposure and mitochondrial epigenetics: Study design and analytical challenges. Hum. Genet. 2014, 133, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Wijst, M.G.; Rots, M.G. Mitochondrial epigenetics: An overlooked layer of regulation? Trends Genet. 2015, 31, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Cuenin, C.; Chung, F.; Aguilera, J.R.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef] [Green Version]
- Dou, X.; Boyd-Kirkup, J.D.; McDermott, J.; Zhang, X.; Li, F.; Rong, B.; Zhang, R.; Miao, B.; Chen, P.; Cheng, H. The strand-biased mitochondrial DNA methylome and its regulation by DNMT3A. Genome Res. 2019, 29, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Sirard, M.-A. Distribution and dynamics of mitochondrial DNA methylation in oocytes, embryos and granulosa cells. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Dostal, V.; Churchill, M.E. Cytosine methylation of mitochondrial DNA at CpG sequences impacts transcription factor A DNA binding and transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Wu, T.; Cui, X.; Zhu, P.; Tan, C.; Dou, X.; Hsu, K.-W.; Lin, Y.-T.; Peng, P.-H.; Zhang, L.-S. N6-Deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell 2020, 78, 382–395.e388. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, P.; Cui, Y.; Wang, Y. Chemical analysis of DNA damage. Anal. Chem. 2018, 90, 556. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y. Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem. Soc. Rev. 2015, 44, 7829–7854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, T.P.; Dolan, M.E.; Fraenkel-Conrat, H.; Hall, J.; Karran, P.; Laval, L.; Margison, G.P.; Montesano, R.; Pegg, A.E.; Potter, P.M. Repair of O-alkylpyrimidines in mammalian cells: A present consensus. Proc. Natl. Acad. Sci. USA 1988, 85, 1759–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronstein, S.M.; Skopek, T.R.; Swenberg, J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992, 52, 2008–2011. [Google Scholar] [PubMed]
- Stepanov, I.; Hecht, S.S. Mitochondrial DNA adducts in the lung and liver of F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and (S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 406–414. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Wang, P.; Wang, Y. Mitochondrial Transcription Factor A Binds to and Promotes Mutagenic Transcriptional Bypass of O 4-Alkylthymidine Lesions. Anal. Chem. 2020, 93, 1161–1169. [Google Scholar] [CrossRef]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The endogenous exposome. DNA Repair 2014, 19, 3–13. [Google Scholar] [CrossRef]
- Greenberg, M.M. Looking beneath the surface to determine what makes DNA damage deleterious. Curr. Opin. Chem. Biol. 2014, 21, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Boyd, R.M.; Tree, M.O.; Samkari, F.; Zhao, L. Mitochondrial transcription factor A promotes DNA strand cleavage at abasic sites. Proc. Natl. Acad. Sci. USA 2019, 116, 17792–17799. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial DNA: A disposable genome? Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1805–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The structure and function of DNA G-quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Dolinnaya, N.G.; Ogloblina, A.M.; Yakubovskaya, M.G. Structure, properties, and biological relevance of the DNA and RNA G-quadruplexes: Overview 50 years after their discovery. Biochemistry Moscow 2016, 81, 1602–1649. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Tseng, T.-Y.; Chen, Y.-T.; Chang, C.-C.; Wang, Z.-F.; Wang, C.-L.; Hsu, T.-N.; Li, P.-T.; Chen, C.-T.; Lin, J.-J. Direct evidence of mitochondrial G-quadruplex DNA by using fluorescent anti-cancer agents. Nucleic Acids Res. 2015, 43, 10102–10113. [Google Scholar] [CrossRef] [Green Version]
- Falabella, M.; Kolesar, J.; Wallace, C.; De Jesus, D.; Sun, L.; Taguchi, Y.; Wang, C.; Wang, T.; Xiang, I.; Alder, J. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci. Rep. 2019, 9, 1–17. [Google Scholar]
- Sullivan, E.D.; Longley, M.J.; Copeland, W.C. Polymerase γ efficiently replicates through many natural template barriers but stalls at the HSP1 quadruplex. J. Biol. Chem. 2020, 295, 17802–17815. [Google Scholar] [CrossRef] [PubMed]
- Falabella, M.; Fernandez, R.J.; Johnson, F.B.; Kaufman, B.A. Potential roles for G-quadruplexes in mitochondria. Curr. Med. Chem. 2019, 26, 2918–2932. [Google Scholar] [CrossRef]
- Lyonnais, S.; Tarrés-Soler, A.; Rubio-Cosials, A.; Cuppari, A.; Brito, R.; Jaumot, J.; Gargallo, R.; Vilaseca, M.; Silva, C.; Granzhan, A. The human mitochondrial transcription factor A is a versatile G-quadruplex binding protein. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.Z.; Zhu, J.; Dagda, R.K.; Uechi, G.; Cherra III, S.J.; Gusdon, A.M.; Balasubramani, M.; Chu, C.T. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: Implications for Parkinson’s disease. Mitochondrion 2014, 17, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem. Sci. 2016, 41, 231–244. [Google Scholar] [CrossRef] [Green Version]
- King, G.A.; Hashemi Shabestari, M.; Taris, K.-K.H.; Pandey, A.K.; Venkatesh, S.; Thilagavathi, J.; Singh, K.; Krishna Koppisetti, R.; Temiakov, D.; Roos, W.H. Acetylation and phosphorylation of human TFAM regulate TFAM–DNA interactions via contrasting mechanisms. Nucleic Acids Res. 2018, 46, 3633–3642. [Google Scholar] [CrossRef]
- Chen, Z.; Cole, P.A. Synthetic approaches to protein phosphorylation. Curr. Opin. Chem. Biol. 2015, 28, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Albaugh, B.N.; Arnold, K.M.; Lee, S.; Denu, J.M. Autoacetylation of the histone acetyltransferase Rtt109. J. Biol. Chem. 2011, 286, 24694–24701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Akimoto, M.; Mayanagi, K.; Hatano, A.; Matsumoto, M.; Matsuda, S.; Yasukawa, T.; Kang, D. Chemical acetylation of mitochondrial transcription factor A occurs on specific lysine residues and affects its ability to change global DNA topology. Mitochondrion 2020, 53, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Fracasso, F.; Marzetti, E.; Calvani, R.; Leeuwenburgh, C.; Russo, F.; Lezza, A.M.S.; Pesce, V. Differences in liver TFAM binding to mtDNA and mtDNA damage between aged and extremely aged rats. Int. J. Mol. Sci. 2019, 20, 2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatahet, Z.; Kow, Y.W.; Purmal, A.A.; Cunningham, R.P.; Wallace, S.S. New substrates for old enzymes. 5-Hydroxy-2′-deoxycytidine and 5-hydroxy-2′-deoxyuridine are substrates for Escherichia coli endonuclease III and formamidopyrimidine DNA N-glycosylase, while 5-hydroxy-2′-deoxyuridine is a substrate for uracil DNA N-glycosylase. J. Biol. Chem. 1994, 269, 18814–18820. [Google Scholar] [PubMed]
- Bhagwat, M.; Gerlt, J.A. 3′- and 5′-strand cleavage reactions catalyzed by the Fpg protein from Escherichia coli occur via successive β-and δ-elimination mechanisms, respectively. Biochemistry 1996, 35, 659–665. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chew, K.; Zhao, L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes 2021, 12, 1246. https://doi.org/10.3390/genes12081246
Chew K, Zhao L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes. 2021; 12(8):1246. https://doi.org/10.3390/genes12081246
Chicago/Turabian StyleChew, Krystie, and Linlin Zhao. 2021. "Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications" Genes 12, no. 8: 1246. https://doi.org/10.3390/genes12081246
APA StyleChew, K., & Zhao, L. (2021). Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes, 12(8), 1246. https://doi.org/10.3390/genes12081246