
Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches

 and
and
Abstract
:1. Introduction
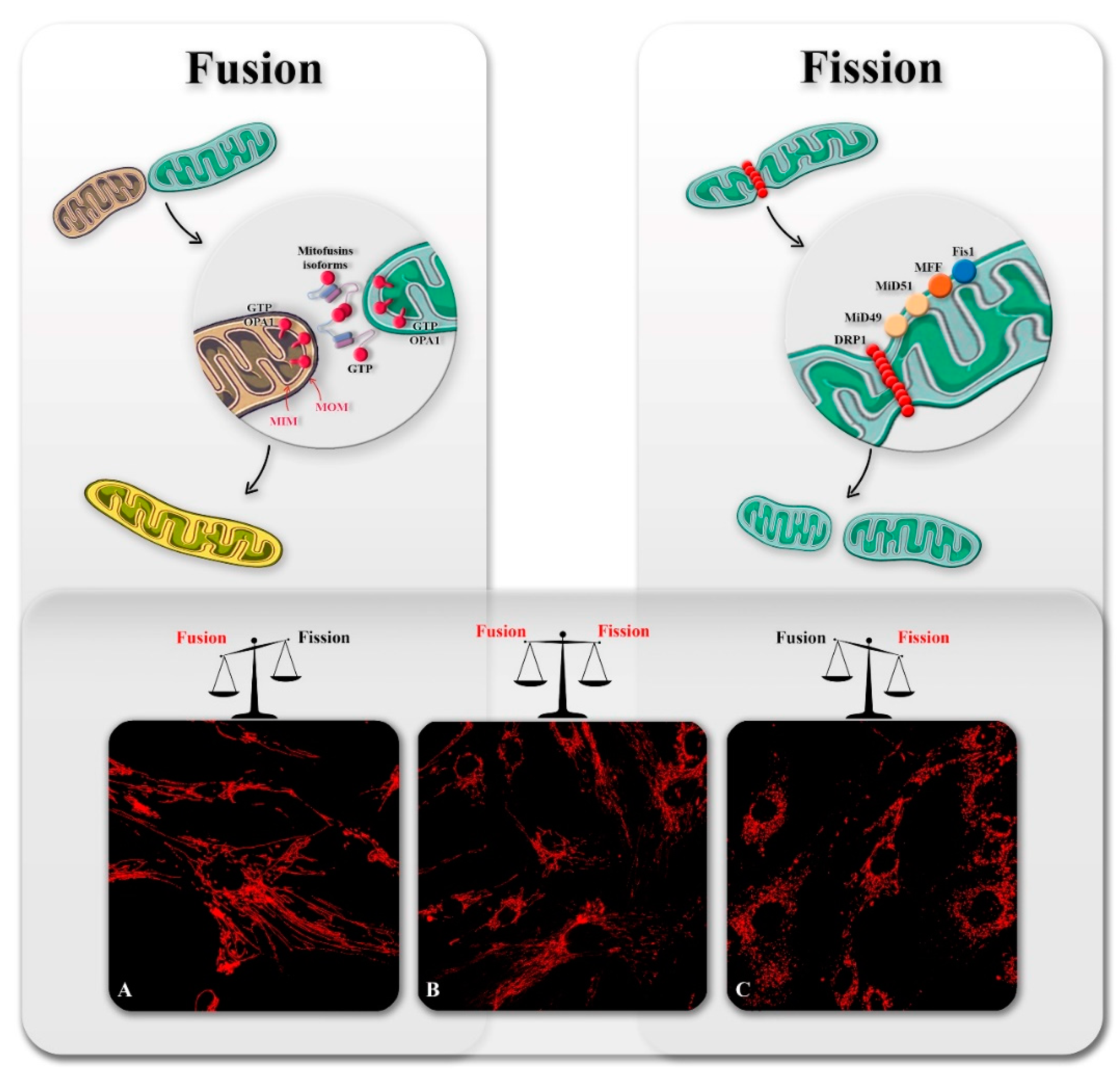
2. Overview of Mitochondrial Dynamics
2.1. Mitochondrial Fission
2.2. Mitochondrial Fusion
3. Mitochondrial Dynamics Related Disorders
3.1. Fission Related Mitochondrial Diseases
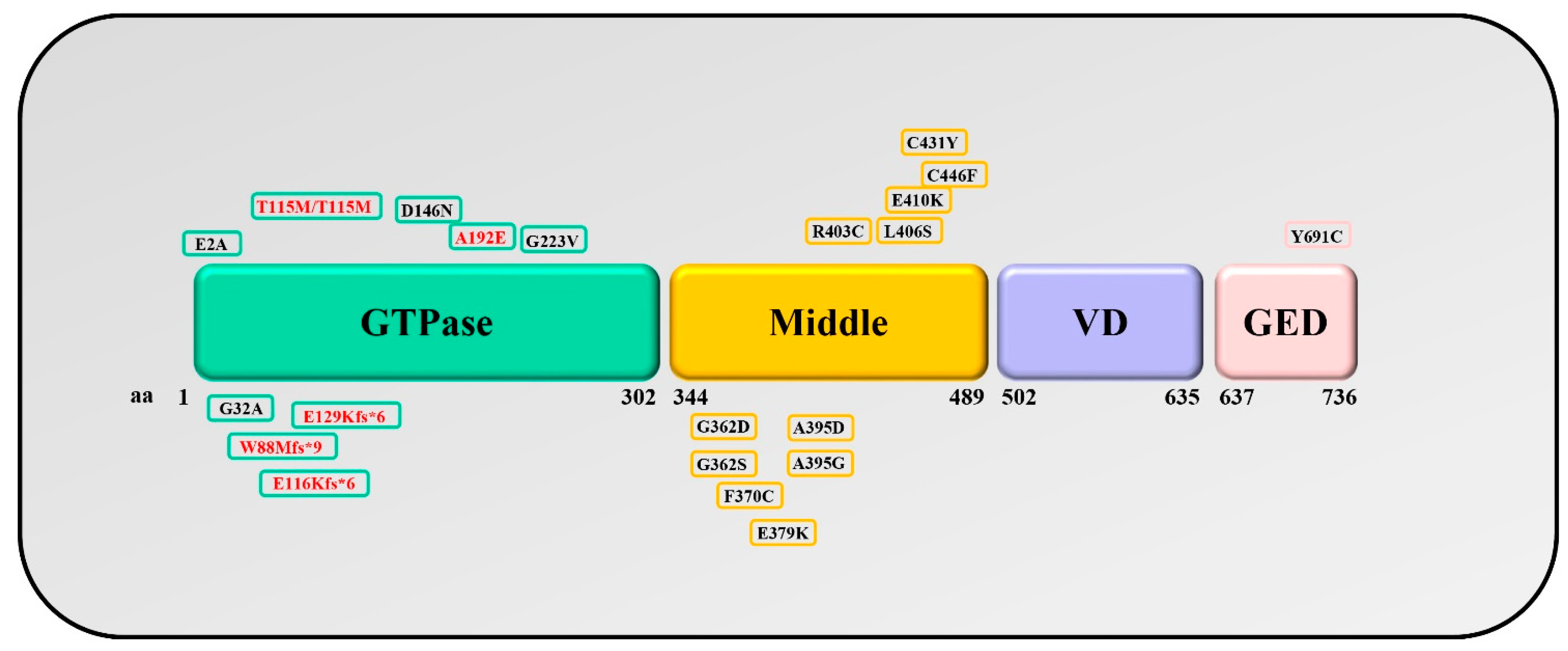
3.1.1. Drp1
3.1.2. Mff
3.1.3. MIDs
3.2. Diseases Related to Mitochondrial Fusion Abnormalities
3.2.1. OPA1
3.2.2. YME1L1
3.2.3. MFN2
3.2.4. MSTO1
3.2.5. FBXL4
4. Therapeutic Approaches
4.1. Correcting DNM1L-Related Defects
4.2. OPA1 Related Therapies
4.3. Therapeutic Perspectives for MFN2-Related Disorders
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dasgupta, S. Mitochondrion: I am more than a fuel server. Ann. Transl. Med. 2019, 7, 594. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Ryan, M.T. Mitochondrial OXPHOS complex assembly lines. Nat. Cell Biol. 2018, 20, 511–513. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Distelmaier, F.; Smeitink, J.A.M.; Willems, P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2012, 32, 9–29. [Google Scholar] [CrossRef]
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 575–587. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M. Mitochondrial disease genetics update. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Munnich, A.; Rötig, A.; Chretien, D.; Cormier, V.; Bourgeron, T.; Bonnefont, J.-P.; Saudubray, J.-M.; Rustin, P. Clinical presentation of mitochondrial disorders in childhood. J. Inherit. Metab. Dis. 1996, 19, 521–527. [Google Scholar] [CrossRef]
- Murayama, K.; Shimura, M.; Liu, Z.; Okazaki, Y.; Ohtake, A. Recent topics: The diagnosis, molecular genesis, and treatment of mitochondrial diseases. J. Hum. Genet. 2018, 64, 113–125. [Google Scholar] [CrossRef]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial dynamics in basal and stressful conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Marín-Hernández, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortés-Hernández, P. Mitochondrial dynamics and cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexiou, A.; Nizami, B.; Khan, F.I.; Soursou, G.; Vairaktarakis, C.; Chatzichronis, S.; Tsiamis, V.; Manztavinos, V.; Yarla, N.S.; Ashraf, G.M.; et al. Mitochondrial dynamics and proteins related to neurodegenerative diseases. Curr. Protein Pept. Sci. 2018, 19, 850–857. [Google Scholar] [CrossRef]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2010, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Francy, C.A.; Alvarez, F.J.D.; Zhou, L.; Ramachandran, R.; Mears, J.A. The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. J. Biol. Chem. 2015, 290, 11692–11703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Gandre-Babbe, S.; Van Der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.A.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Losón, O.C.; Meng, S.; Ngo, H.; Liu, R.; Kaiser, J.T.; Chan, D.C. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. 2015, 24, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, V.; Palmer, C.S.; Osellame, L.D.; Singh, A.P.; Elgass, K.; Stroud, D.A.; Sesaki, H.; Kvansakul, M.; Ryan, M.T. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J. Cell Biol. 2014, 204, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekkes, P.; Shepard, K.A.; Yaffe, M.P. Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J. Cell Biol. 2000, 151, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Mozdy, A.; McCaffery, J.; Shaw, J. Dnm1p Gtpase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [Green Version]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [Green Version]
- Stojanovski, D.; Koutsopoulos, O.S.; Okamoto, K.; Ryan, M.T. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 2004, 117, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [Green Version]
- Rojansky, R.; Cha, M.-Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 2016, 5, e17896. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 2014, 24, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Sweitzer, S.M.; Hinshaw, J.E. Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 1998, 93, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Fonseca, T.B.; Sánchez-Guerrero, Á.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Taguchi, T.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K.; Nakagawa, T.; Uozumi, N.; Nakano, M.; Mizuno-Horikawa, Y.; Okuyama, N.; et al. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Cereghetti, G.M.; Stangherlin, A.; De Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [Green Version]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R.; Hoshijima, M.; Dillmann, W.H. Modulation of dynamin-related protein 1 (DRP1) Function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 2012, 287, 30024–30034. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikstrom, J.D.; Twig, G.; Shirihai, O.S. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 2009, 41, 1914–1927. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Long, Q.; Liu, J.; Tang, H.; Li, Y.; Bao, F.; Qin, D.; Pei, D.; Liu, X. Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell. Mol. Life Sci. 2015, 72, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnóczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion–fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Cohen, M.M.; Tareste, D. Recent insights into the structure and function of Mitofusins in mitochondrial fusion. F1000Research 2018, 7, 1983. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, T. Structural basis of mitochondrial tethering by Mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daste, F.; Sauvanet, C.; Bavdek, A.; Baye, J.; Pierre, F.; Le Borgne, R.; David, C.; Rojo, M.; Fuchs, P.; Tareste, D. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 2018, 19, e43637. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human Mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the miro/milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012, 32, 4145–4155. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.-J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 2018, 175, 1430–1442.e17. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Zunino, R.; Gangaraju, S.; Rippstein, P.; McBride, H. Activated Mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 2005, 280, 25060–25070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.-P.; et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 2004, 6, 872–883. [Google Scholar] [CrossRef]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. 2017, 93, 933–949. [Google Scholar] [CrossRef] [Green Version]
- Schrepfer, E.; Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.-Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated Mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 2014, 28, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Okano, Y. Human Misato regulates mitochondrial distribution and morphology. Exp. Cell Res. 2007, 313, 1393–1404. [Google Scholar] [CrossRef]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Várnai, P.; Gyuris, T.; Horváth, A.; Nagy, L.; Seifert, E.L.; et al. MSTO 1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017, 9, 967–984. [Google Scholar] [CrossRef]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.; Kashiwagi, Y.; Arimitsu, N.; Kogure, T.; Edo, A.; Maruyama, T.; Nakao, K.; Nakanishi, H.; Kinoshita, M.; Frohman, M.A.; et al. Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J. Biol. Chem. 2014, 289, 11497–11511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belenguer, P.; Pellegrini, L. The dynamin GTPase OPA1: More than mitochondria? Biochim. Biophys. Acta 2013, 1833, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; ElAchouri, G.; Baricault, L.; Delettre, C.; Belenguer, P.; Lenaers, G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 2006, 14, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.; Hamamoto, T.; Seo, N.; Kagawa, Y.; Endo, H. Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem. Biophys. Res. Commun. 2003, 300, 482–493. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; Van Der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Griparic, L.; Kanazawa, T.; Van Der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA 1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Löwer, B.; Wunderlich, F.T.; Von Kleist-Retzow, J.-C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, T.; Kohno, H.; Ishihara, T.; Ishihara, N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9. [Google Scholar] [CrossRef]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S.; et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 2013, 93, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Antoun, G.; McBride, S.; Vanstone, J.R.; Naas, T.; Michaud, J.; Redpath, S.; McMillan, H.J.; Brophy, J.; Daoud, H.; Chakraborty, P.; et al. Detailed biochemical and bioenergetic characterization of FBXL4-related encephalomyopathic mitochondrial DNA depletion. JIMD Rep. 2015, 27, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sabouny, R.; Wong, R.; Lee-Glover, L.; Greenway, S.C.; Sinasac, D.S.; Khan, A.; Shutt, T.E. Characterization of the C584R variant in the mtDNA depletion syndrome gene FBXL4, reveals a novel role for FBXL4 as a regulator of mitochondrial fusion. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 165536. [Google Scholar] [CrossRef]
- Park, Y.-Y.; Nguyen, O.T.K.; Kang, H.; Cho, H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kapur, M.; Li, M.; Choi, M.-C.; Choi, S.; Kim, H.-J.; Kim, I.; Lee, E.; Taylor, J.P.; Yao, T.-P. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 2014, 127, 4954–4963. [Google Scholar] [CrossRef] [Green Version]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870, Erratum in 2013, 22, 1697. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; Van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.-E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef]
- Chao, Y.-H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Zaha, K.; Matsumoto, H.; Itoh, M.; Saitsu, H.; Kato, K.; Kato, M.; Ogata, S.; Murayama, K.; Kishita, Y.; Mizuno, Y.; et al. DNM1L-related encephalopathy in infancy with Leigh syndrome-like phenotype and suppression-burst. Clin. Genet. 2016, 90, 472–474. [Google Scholar] [CrossRef]
- Díez, H.; Cortès-Saladelafont, E.; Ormazábal, A.; Marmiese, A.F.; Armstrong, J.; Matalonga, L.; Bravo, M.; Briones, P.; Emperador, S.; Montoya, J.; et al. Severe infantile parkinsonism because of a de novo mutation on DLP1 mitochondrial-peroxisomal protein. Mov. Disord. 2017, 32, 1108–1110. [Google Scholar] [CrossRef]
- Ryan, C.S.; Fine, A.L.; Cohen, A.L.; Schiltz, B.M.; Renaud, D.L.; Wirrell, E.C.; Patterson, M.C.; Boczek, N.J.; Liu, R.; Babovic-Vuksanovic, D.; et al. De Novo DNM1L variant in a teenager with progressive paroxysmal dystonia and lethal super-refractory myoclonic status epilepticus. J. Child. Neurol. 2018, 33, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Ladds, E.; Whitney, A.; Dombi, E.; Hofer, M.; Anand, G.; Harrison, V.; Fratter, C.; Carver, J.; Barbosa, I.A.; Simpson, M.A.; et al. De novo DNM1L mutation associated with mitochondrial epilepsy syndrome with fever sensitivity. Neurol. Genet. 2018, 4, e258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, B.N.; Lam, C.; Cui, H.; Haude, K.; Bai, R.; Escobar, L.; Hamilton, A.; Brady, L.; Tarnopolsky, M.A.; Dengle, L.; et al. Aberrant Drp1-mediated mitochondrial division presents in humans with variable outcomes. Hum. Mol. Genet. 2018, 27, 3710–3719. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.A.; Chen, B.; Michon, A.M.; Salatka, E.; Arndt, D. A Rasmussen encephalitis, autoimmune encephalitis, and mitochondrial disease mimicker: Expanding the DNM1L-associated intractable epilepsy and encephalopathy phenotype. Epileptic Disord. 2019, 21, 112–116. [Google Scholar] [PubMed]
- Verrigni, D.; Di Nottia, M.; Ardissone, A.; Baruffini, E.; Nasca, A.; Legati, A.; Bellacchio, E.; Fagiolari, G.; Martinelli, D.; Fusco, L.; et al. Clinical-genetic features and peculiar muscle histopathology in infantile DNM1L-related mitochondrial epileptic encephalopathy. Hum. Mutat. 2019, 40, 601–618. [Google Scholar] [CrossRef]
- Schmid, S.J.; Wagner, M.; Goetz, C.; Makowski, C.; Freisinger, P.; Berweck, S.; Mall, V.; Burdach, S.; Juenger, H. A De Novo dominant negative mutation in DNM1L causes sudden onset status epilepticus with subsequent epileptic encephalopathy. Neuropediatrics 2019, 50, 197–201. [Google Scholar] [CrossRef]
- Vandeleur, D.; Chen, C.V.; Huang, E.J.; Connolly, A.J.; Sanchez, H.; Moon-Grady, A. Novel and lethal case of cardiac involvement in DNM1L mitochondrial encephalopathy. Am. J. Med. Genet. A 2019, 179, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Zahir, F.R.; Zivkovic, I.; Moksa, M.; Selby, K.; Sinha, S.; Nislow, C.; Stockler-Ipsiroglu, S.G.; Sheffer, R.; Saada-Reisch, A.; et al. De novo pathogenic DNM1L variant in a patient diagnosed with atypical hereditary sensory and autonomic neuropathy. Mol. Genet. Genom. Med. 2019, 7, e00961. [Google Scholar] [CrossRef] [Green Version]
- McCormack, M.; McGinty, R.N.; Zhu, X.; Slattery, L.; Heinzen, E.L.; Costello, D.J.; Delanty, N.; Cavalleri, G.L.; The EpiGen Consortium. De-novo mutations in patients with chronic ultra-refractory epilepsy with onset after age five years. Eur. J. Med. Genet. 2020, 63, 103625. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, S.; Paris, A.; Alban, J.; Quilès, M.; Delettre, C.; et al. Reply: The expanding neurological phenotype of DNM1L-related disorders. Brain 2018, 141, e29. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Benedetti, S.; Zambon, A.A.; Sora, M.G.N.; Di Resta, C.; De Ritis, D.; Quattrini, A.; Maltecca, F.; Ferrari, M.; Previtali, S.C. Impaired turnover of hyperfused mitochondria in severe axonal neuropathy due to a novel DRP1 mutation. Hum. Mol. Genet. 2019, 29, 177–188. [Google Scholar] [CrossRef]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.-S.; Hawkins, C.; Cohn, R.D.; et al. Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J. Pediatr. 2016, 171, 313–316.e2. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarth, K.A.; Costford, S.R.; Yoon, G.; Sondheimer, N.; Maynes, J.T. DNM1L variant alters baseline mitochondrial function and response to stress in a patient with severe neurological dysfunction. Biochem. Genet. 2017, 56, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Batzir, N.A.; Bhagwat, P.K.; Eble, T.N.; Liu, P.; Eng, C.M.; Elsea, S.H.; Robak, L.A.; Scaglia, F.; Goldman, A.M.; Dhar, S.U.; et al. De novo missense variant in the GTPase effector domain (GED) of DNM1L leads to static encephalopathy and seizures. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Shamseldin, H.E.; Alshammari, M.; Al-Sheddi, T.; Salih, M.A.; Alkhalidi, H.; Kentab, A.; Repetto, G.M.; Hashem, M.; Alkuraya, F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012, 49, 234–241. [Google Scholar] [CrossRef]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Nardecchia, F.; Commone, A.; Semeraro, M.; Legati, A.; Garavaglia, B.; Ghezzi, D.; Leuzzi, V. Clinical and biochemical features in a patient with mitochondrial fission factor gene alteration. Front. Genet. 2018, 9, 625. [Google Scholar] [CrossRef] [Green Version]
- Panda, I.; Ahmad, I.; Sagar, S.; Zahra, S.; Shamim, U.; Sharma, S.; Faruq, M. Encephalopathy due to defective mitochondrial and peroxisomal fission 2 caused by a novel MFF gene mutation in a young child. Clin. Genet. 2020, 97, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Bartsakoulia, M.; Pyle, A.; Troncoso-Chandía, D.; Vial-Brizzi, J.; Paz-Fiblas, M.V.; Duff, J.; Griffin, H.; Boczonadi, V.; Lochmüller, H.; Kleinle, S.; et al. A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Hum. Mol. Genet. 2018, 27, 1186–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilz, Y.L.; Bass, S.J.; Sherman, J. A review of mitochondrial optic neuropathies: From inherited to acquired forms. J. Optom. 2017, 10, 205–214. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; De La Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2007, 131, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Shankar, S.P.; Biousse, V.; Miller, N.R.; Bean, L.J.; Coffee, B.; Hegde, M.; Newman, N.J. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology 2011, 118, 558–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresca, A.; La Morgia, C.; Caporali, L.; Valentino, M.L.; Carelli, V. The optic nerve: A “mito-window” on mitochondrial neurodegeneration. Mol. Cell. Neurosci. 2013, 55, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Pesch, U.E.A.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, C.; Blazo, M.; Mann, J.; Tonini, R.E.; Takei, H.; Wang, J.; Wong, L.-J.C.; Scaglia, F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011, 103, 383–387. [Google Scholar] [CrossRef]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [Green Version]
- Carelli, V.; Sabatelli, M.; Carrozzo, R.; Rizza, T.; Schimpf, S.; Wissinger, B.; Zanna, C.; Rugolo, M.; La Morgia, C.; Caporali, L.; et al. ‘Behr syndrome’ with OPA1 compound heterozygote mutations. Brain 2015, 138, e321. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jung, S.-C.; Bin Hong, Y.; Yoo, J.H.; Koo, H.; Lee, J.H.; Hong, H.D.; Kim, S.-B.; Chung, K.W.; Choi, B.-O. Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1. Mol. Med. Rep. 2016, 14, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Rubegni, A.; Pisano, T.; Bacci, G.; Tessa, A.; Battini, R.; Procopio, E.; Giglio, S.; Pasquariello, R.; Santorelli, F.M.; Guerrini, R.; et al. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur. J. Paediatr. Neurol. 2017, 21, 671–677. [Google Scholar] [CrossRef]
- Nasca, A.; Rizza, T.; Doimo, M.; Legati, A.; Andrea, C.; Diodato, N.; Calderan, C.; Carrara, G.; Lamantea, E.; Aiello, C.; et al. Not only dominant, not only optic atrophy: Expanding the clinical spectrum associated with OPA1 mutations. Orphanet J. Rare Dis. 2017, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerem, A.; Yosovich, K.; Rappaport, Y.C.; Libzon, S.; Blumkin, L.; Ben-Sira, L.; Lev, D.; Lerman-Sagie, T. Metabolic stroke in a patient with bi-allelic OPA1 mutations. Metab. Brain Dis. 2019, 34, 1043–1048. [Google Scholar] [CrossRef]
- Bonifert, T.; Karle, K.N.; Tonagel, F.; Batra, M.; Wilhelm, C.; Theurer, Y.; Schoenfeld, C.; Kluba, T.; Kamenisch, Y.; Carelli, V.; et al. Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 2014, 137, 2164–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, B.; Wai, T.; Hu, H.; MacVicar, T.; Musante, L.; Fischer-Zirnsak, B.; Stenzel, W.; Gräf, R.; Heuvel, L.V.D.; Ropers, H.-H.; et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 2016, 5, e16078. [Google Scholar] [CrossRef] [PubMed]
- Barreto, L.C.L.S.; Oliveira, F.S.; Nunes, P.S.; de França Costa, I.M.P.; Garcez, C.A.; Goes, G.M.; Neves, E.L.A.; de Souza Siqueira Quintans, J.; de Souza Araújo, A.A. Epidemiologic study of Charcot-Marie-Tooth disease: A systematic review. Neuroepidemiology 2016, 46, 157–165. [Google Scholar] [CrossRef]
- Cortese, A.; Wilcox, J.E.; Polke, J.M.; Poh, R.; Skorupinska, M.; Rossor, A.M.; Laura, M.; Tomaselli, P.J.; Houlden, H.; Shy, M.E.; et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 2020, 94, e51–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.-F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Bombelli, F.; Stojkovic, T.; Dubourg, O.; Echaniz-Laguna, A.; Tardieu, S.; Larcher, K.; Amati-Bonneau, P.; Latour, P.; Vignal, O.; Cazeneuve, C.; et al. Charcot-Marie-Tooth disease type 2A. JAMA Neurol. 2014, 71, 1036–1042. [Google Scholar] [CrossRef]
- Leonardi, L.; Marcotulli, C.; Storti, E.; Tessa, A.; Serrao, M.; Parisi, V.; Santorelli, F.M.; Pierelli, F.; Casali, C. Acute optic neuropathy associated with a novel MFN2 mutation. J. Neurol. 2015, 262, 1678–1680. [Google Scholar] [CrossRef]
- Ando, M.; Hashiguchi, A.; Okamoto, Y.; Yoshimura, A.; Hiramatsu, Y.; Yujiro, H.; Higuchi, Y.; Mitsui, J.; Ishiura, H.; Umemura, A.; et al. Clinical and genetic diversities of Charcot-Marie-Tooth disease with MFN2 mutations in a large case study. J. Peripher. Nerv. Syst. 2017, 22, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, S.; D’Oria, F.; Rossetti, G.; Favale, R.A.; Zoccolella, S.; Alessio, G.; Petruzzella, V. CMT2A harboring Mitofusin 2 mutation with optic nerve atrophy and normal visual acuity. Int. Med. Case Rep. J. 2020, 13, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, K.; Claeys, K.G.; Züchner, S.; Schröder, J.M.; Weis, J.; Ceuterick, C.; Jordanova, A.; Nelis, E.; De Vriendt, E.; Van Hul, M.; et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 2006, 129, 2093–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, G.A.; Magdelaine, C.; Zhu, D.; Grew, S.; Ryan, M.M.; Sturtz, F.; Vallat, J.-M.; Ouvrier, R.A. Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology 2008, 70, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Vallat, J.-M.; Ouvrier, R.A.; Pollard, J.D.; Magdelaine, C.; Zhu, D.; Nicholson, G.A.; Grew, S.; Ryan, M.M.; Funalot, B. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with Mitofusin 2 mutations. J. Neuropathol. Exp. Neurol. 2008, 67, 1097–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, J.; Funalot, B.; Ouvrier, R.A.; Lazaro, L.; Toutain, A.; De Mas, P.; Bouche, P.; Gilbert-Dussardier, B.; Arne-Bes, M.-C.; Carrière, J.-P.; et al. Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by Mitofusin 2 mutations. Arch. Neurol. 2009, 66, 1511–1516. [Google Scholar] [CrossRef]
- Polke, J.M.; Laura, M.; Pareyson, D.; Taroni, F.; Milani, M.; Bergamin, G.; Gibbons, V.S.; Houlden, H.; Chamley, S.C.; Blake, J.; et al. Recessive axonal Charcot-Marie-Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology 2011, 77, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, C.; Trajanoski, S.; Papić, L.; Windpassinger, C.; Bernert, G.; Freilinger, M.; Schabhüttl, M.; Arslan-Kirchner, M.; Javaher-Haghighi, P.; Plecko, B.; et al. SNP array-based whole genome homozygosity mapping as the first step to a molecular diagnosis in patients with Charcot-Marie-Tooth disease. J. Neurol. 2012, 259, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.S.; Polke, J.M.; Wilson, J.; Pelayo-Negro, A.L.; Laura, M.; Nanji, T.; Holt, J.B.; Vaughan, J.; Rankin, J.; Sweeney, M.G.; et al. MFN2 deletion of exons 7 and 8: Founder mutation in the UK population. J. Peripher. Nerv. Syst. 2015, 20, 67–71. [Google Scholar] [CrossRef]
- Piscosquito, G.; Saveri, P.; Magri, S.; Ciano, C.; Di Bella, D.; Milani, M.; Taroni, F.; Pareyson, D. Mutational mechanisms in MFN2-related neuropathy: Compound heterozygosity for recessive and semidominant mutations. J. Peripher. Nerv. Syst. 2015, 20, 380–386. [Google Scholar] [CrossRef]
- Tan, C.A.; Rabideau, M.; Blevins, A.; Westbrook, M.J.; Ekstein, T.; Nykamp, K.; Deucher, A.; Harper, A.; Demmer, L. Autosomal recessive MFN2-related Charcot-Marie-Tooth disease with diaphragmatic weakness: Case report and literature review. Am. J. Med. Genet. A 2016, 170, 1580–1584. [Google Scholar] [CrossRef]
- Geroldi, A.; Lastella, P.; Patruno, M.; Gotta, F.; Resta, N.; Devigili, G.; Sabbà, C.; Gulli, R.; Lamp, M.; Origone, P.; et al. Two novel cases of compound heterozygous mutations in mitofusin2: Finding out the inheritance. Neuromuscul. Disord. 2017, 27, 377–381. [Google Scholar] [CrossRef]
- Hikiami, R.; Yamashita, H.; Koita, N.; Jingami, N.; Sawamoto, N.; Furukawa, K.; Kawai, H.; Terashima, T.; Oka, N.; Hashiguchi, A.; et al. Charcot–Marie–Tooth disease type 2A with an autosomal-recessive inheritance: The first report of an adult-onset disease. J. Hum. Genet. 2017, 63, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Iapadre, G.; Morana, G.; Vari, M.S.; Pinto, F.; Lanteri, P.; Tessa, A.; Santorelli, F.M.; Striano, P.; Verrotti, A. A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur. J. Paediatr. Neurol. 2018, 22, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Stuppia, G.; Rizzo, F.; Riboldi, G.; Del Bo, R.; Nizzardo, M.; Simone, C.; Comi, G.P.; Bresolin, N.; Corti, S. MFN2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J. Neurol. Sci. 2015, 356, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Iwama, K.; Takaori, T.; Fukushima, A.; Tohyama, J.; Ishiyama, A.; Ohba, C.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N.; et al. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J. Hum. Genet. 2018, 63, 263–270. [Google Scholar] [CrossRef]
- Li, K.; Jin, R.; Wu, X. Whole-exome sequencing identifies rare compound heterozygous mutations in the MSTO1 gene associated with cerebellar ataxia and myopathy. Eur. J. Med. Genet. 2020, 63, 103623. [Google Scholar] [CrossRef] [PubMed]
- Ardicli, D.; Sarkozy, A.; Zaharieva, I.; Deshpande, C.; Bodi, I.; Siddiqui, A.; U-King-Im, J.M.; Selfe, A.; Phadke, R.; Jungbluth, H.; et al. A novel case of MSTO1 gene related congenital muscular dystrophy with progressive neurological involvement. Neuromuscul. Disord. 2019, 29, 448–455. [Google Scholar] [CrossRef]
- Donkervoort, S.; Care4Rare Canada Consortium; Sabouny, R.; Yun, P.; Gauquelin, L.; Chao, K.R.; Hu, Y.; Al Khatib, I.; Töpf, A.; Mohassel, P.; et al. MSTO1 mutations cause mtDNA depletion, manifesting as muscular dystrophy with cerebellar involvement. Acta Neuropathol. 2019, 138, 1013–1031. [Google Scholar] [CrossRef] [Green Version]
- Huemer, M.; Karall, D.; Schossig, A.; Abdenur, J.E.; Al Jasmi, F.; Biagosch, C.; Distelmaier, F.; Freisinger, P.; Graham, B.H.; Haack, T.B.; et al. Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations. J. Inherit. Metab. Dis. 2015, 38, 905–914. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; Heuvel, L.V.D.; Rodenburg, R. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Van Rij, M.C.; Jansen, F.A.R.; Hellebrekers, D.M.E.I.; Onkenhout, W.; Smeets, H.J.M.; Hendrickx, A.T.; Gottschalk, R.W.H.; Steggerda, S.J.; Peeters-Scholte, C.M.P.C.D.; Haak, M.C.; et al. Polyhydramnios and cerebellar atrophy: A prenatal presentation of mitochondrial encephalomyopathy caused by mutations in the FBXL4 gene. Clin. Case Rep. 2016, 4, 425–428. [Google Scholar] [CrossRef] [Green Version]
- Barøy, T.; Pedurupillay, C.R.J.; Bliksrud, Y.T.; Rasmussen, M.; Holmgren, A.; Vigeland, M.D.; Hughes, T.; Brink, M.; Rodenburg, R.; Nedregaard, B.; et al. A novel mutation in FBXL4 in a Norwegian child with encephalomyopathic mitochondrial DNA depletion syndrome. Eur. J. Med. Genet. 2016, 59, 342–346. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Zhang, V.W.; El-Hattab, A.W.; Ficicioglu, C.; Shinawi, F.M.; Lines, M.; Schulze, A.; McNutt, M.; Gotway, G.; Tian, X.; et al. FBXL4 defects are common in patients with congenital lactic acidemia and encephalomyopathic mitochondrial DNA depletion syndrome. Clin. Genet. 2016, 91, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Neilan, E.G.; Peake, R.W.A.; Shi, J.; Schmitz-Abe, K.; Towne, M.; Markianos, K.; Prabhu, S.P.; Agrawal, P.B.; Baumgartner, M.R.; et al. Hyperammonemia as a presenting feature in two siblings with FBXL4 variants. JIMD Rep. 2016, 35, 7–15. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Dai, H.; Almannai, M.; Wang, J.; Faqeih, E.A.; Al Asmari, A.; Saleh, M.A.M.; Elamin, M.A.O.; Alfadhel, M.; Alkuraya, F.S.; et al. Molecular and clinical spectra of FBXL4 deficiency. Hum. Mutat. 2017, 38, 1649–1659. [Google Scholar] [CrossRef]
- Kuptanon, C.; Srichomthong, C.; Ittiwut, C.; Wechapinan, T.; Sri-Udomkajorn, S.; Iamopas, O.; Phokaew, C.; Suphapeetiporn, K.; Shotelersuk, V. Whole exome sequencing revealed mutations in FBXL4, UNC80, and ADK in Thai patients with severe intellectual disabilities. Gene 2019, 696, 21–27. [Google Scholar] [CrossRef]
- Ballout, R.A.; Al Alam, C.; Bonnen, P.E.; Huemer, M.; El-Hattab, A.W.; Shbarou, R. FBXL4-related mitochondrial DNA depletion syndrome 13 (MTDPS13): A case report with a comprehensive mutation review. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emperador, S.; Garrido-Pérez, N.; Amezcua-Gil, J.; Gaudó, P.; Andrés-Sanz, J.A.; Yubero, D.; Fernández-Marmiesse, A.; O’Callaghan, M.M.; Ortigoza-Escobar, J.D.; Iriondo, M.; et al. Molecular characterization of new FBXL4 mutations in patients with mtDNA depletion syndrome. Front. Genet. 2020, 10, 1300. [Google Scholar] [CrossRef]
- Nardi, N.; Proulx, F.; Brunel-Guiton, C.; Oligny, L.L.; Piché, N.; Mitchell, G.A.; Joyal, J.-S. Fulminant necrotizing enterocolitis and multiple organ dysfunction in a toddler with mitochondrial DNA depletion syndrome-13. J. Pediatr. Intensiv. Care 2019, 9, 54–59. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Seveno, M.; Piro-Mégy, C.; Elzière, L.; Quilès, M.; Péquignot, M.; Müller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci. Rep. 2018, 8, 2468. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Cabrera-Pérez, R.; Vila-Julià, F.; Viscomi, C.; Cámara, Y.; Hirano, M.; Zeviani, M.; Martí, R. Long-term sustained effect of liver-targeted adeno-associated virus gene therapy for mitochondrial neurogastrointestinal encephalomyopathy. Hum. Gene Ther. 2018, 29, 708–718. [Google Scholar] [CrossRef]
- Di Meo, I.; Auricchio, A.; Lamperti, C.; Burlina, A.; Viscomi, C.; Zeviani, M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol. Med. 2012, 4, 1008–1014. [Google Scholar] [CrossRef]
- Di Meo, I.; Marchet, S.; Lamperti, C.; Zeviani, M.; Viscomi, C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017, 24, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Karaarslan, C. Leber’s hereditary optic neuropathy as a promising disease for gene therapy development. Adv. Ther. 2019, 36, 3299–3307. [Google Scholar] [CrossRef] [Green Version]
- Douiev, L.; Sheffer, R.; Horvath, G.; Golubitzky, A. Bezafibrate improves mitochondrial fission and function in DNM1L-deficient patient cells. Cells 2020, 9, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.R.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 2012, CD004426. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Amore, G.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 2020, 7, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Aleo, S.J.; Del Dotto, V.; Fogazza, M.; Maresca, A.; Lodi, T.; Goffrini, P.; Ghelli, A.; Rugolo, M.; Carelli, V.; Baruffini, E.; et al. Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum. Mol. Genet. 2021, 29, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djouadi, F.; Bastin, J. Mitochondrial genetic disorders: Cell signaling and pharmacological therapies. Cells 2019, 8, 289. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P. Therapeutic approaches to inherited optic neuropathies. Semin. Neurol. 2015, 35, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2007, 131, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetlin, R.D.; Gutmann, L.; Tarnopolsky, M.; Ullrich, I.H.; Yeater, R.A. Resistance training effectiveness in patients with Charcot-Marie-Tooth disease: Recommendations for exercise prescription. Arch. Phys. Med. Rehabil. 2004, 85, 1217–1223. [Google Scholar] [CrossRef]
- Shy, M.E. Therapeutic strategies for the inherited neuropathies. Neuromolecular Med. 2006, 8, 255–278. [Google Scholar] [CrossRef]
- Herrmann, D.N. Experimental therapeutics in hereditary neuropathies: The past, the present, and the future. Neurotherapeutics 2008, 5, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.-G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, A.H.; Meng, S.; Chu, Q.N.; Chan, D.C. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 2012, 21, 4817–4826. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Small Molecules | |||
|---|---|---|---|
| Genes Related-Disease | Treatment | Outcome | Reference |
| DNM1L | bezafibrate | Improvement of growth, adenosine triphosphate (ATP) production, and oxygen consumption in patient’s fibroblasts harboring the p.G362S variant in the DNM1L gene | [197] |
| OPA1 | coenzyme Q10 | No evidence for a significant benefit in dominant optic atrophy (DOA) patients | [198] |
| idebenone | In 74 of 87 DOA treated patients, an increased visual acuity was observed after at least 7 months of administration | [199] | |
| tolfenamic acid | Positive effects on mtDNA instability energetic metabolism and/or mitochondrial morphology in the yeast model, Opa1 deleted MEFs and patient’ fibroblasts | [200] | |
| MFN2 | mitofusin agonists | Recovery of mitochondrial dysmotility and fragmentation in cultured MFN2 mutated neurons and normalization of the axonal mitochondrial trafficking within sciatic nerves in MFN2T105M transgenic mice | [201] |
| Gene therapy | |||
| OPA1 | AAV2 serotype 2 injection | Positive effects on the electrophysiological measurements specific of retinal ganglion cell activity in Opa1+/− treated mice, but the slow improvement of visual acuity | [192] |
| MFN2 | Thy1.2-MFN2R94Q transgenic mice and Thy1.2-MFN2R94Q:Prp-MFN1 double-transgenic animals | Restoration of MFN1:MFN2 balance by augmenting levels of MFN1 in the nervous system showed near complete rescue of ocular, neuromuscular, and histologic phenotypes | [201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Nottia, M.; Verrigni, D.; Torraco, A.; Rizza, T.; Bertini, E.; Carrozzo, R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes 2021, 12, 247. https://doi.org/10.3390/genes12020247
Di Nottia M, Verrigni D, Torraco A, Rizza T, Bertini E, Carrozzo R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes. 2021; 12(2):247. https://doi.org/10.3390/genes12020247
Chicago/Turabian StyleDi Nottia, Michela, Daniela Verrigni, Alessandra Torraco, Teresa Rizza, Enrico Bertini, and Rosalba Carrozzo. 2021. "Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches" Genes 12, no. 2: 247. https://doi.org/10.3390/genes12020247
APA StyleDi Nottia, M., Verrigni, D., Torraco, A., Rizza, T., Bertini, E., & Carrozzo, R. (2021). Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes, 12(2), 247. https://doi.org/10.3390/genes12020247