
Treacher Collins Syndrome: Genetics, Clinical Features and Management



Abstract
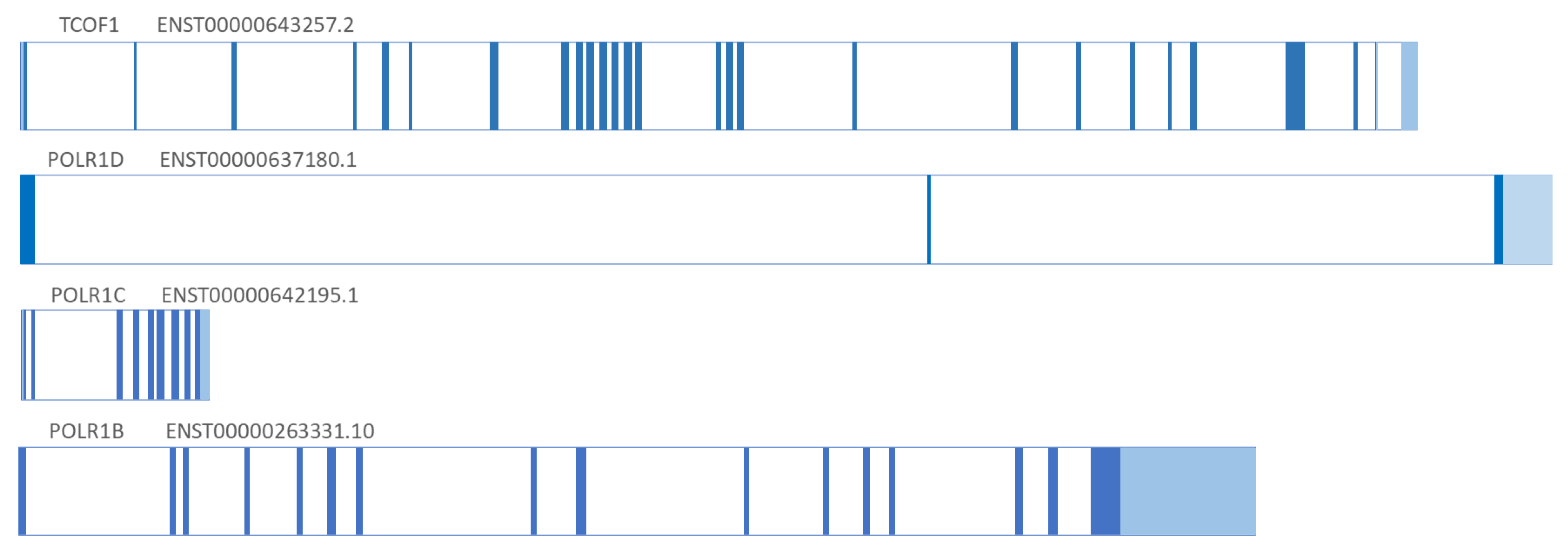
:1. Genetics
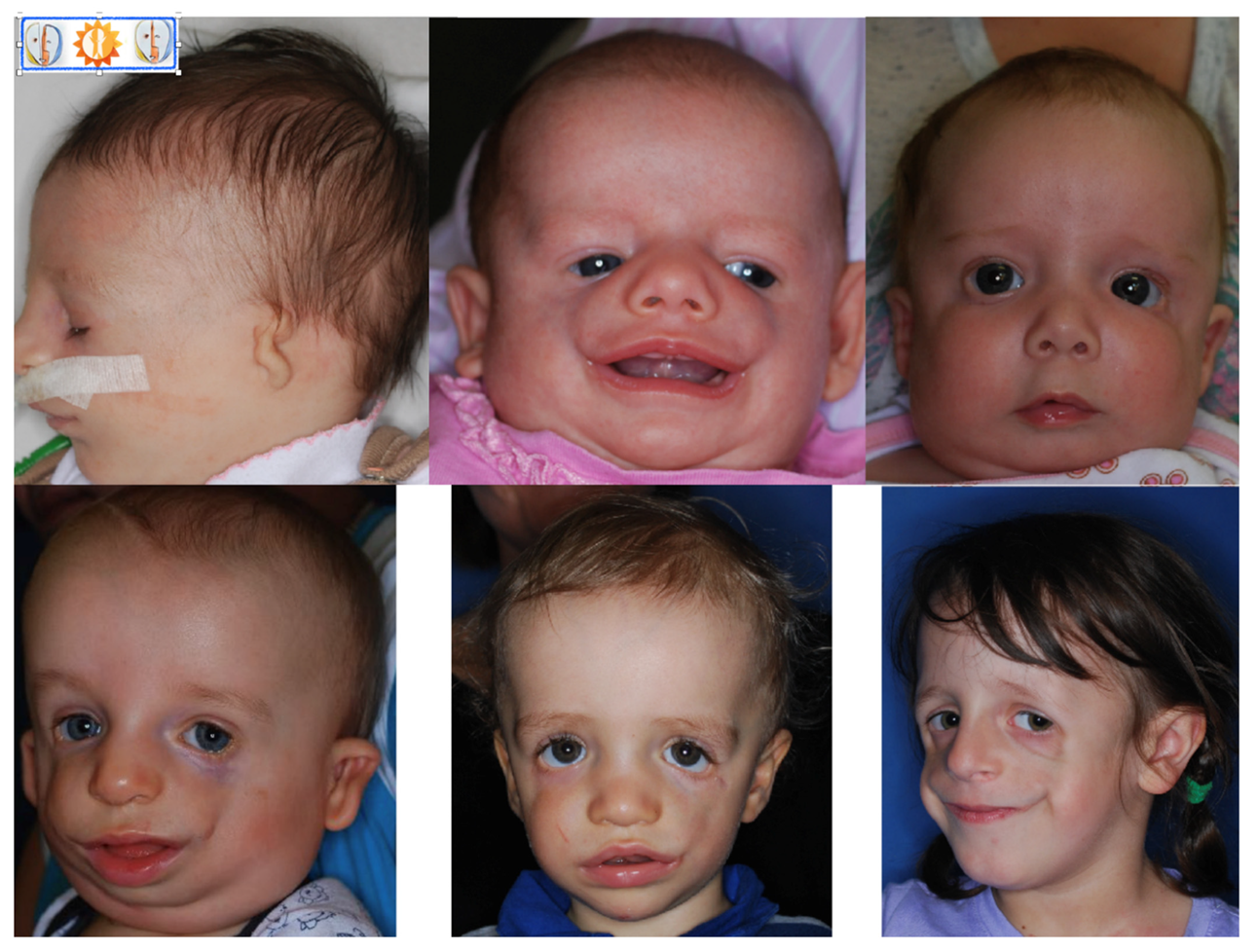
2. Clinical Features–Phenotype
- external and middle ear abnormalities including microtia with conductive hearing loss attributed most commonly to malformation of the ossicles,
- bilateral and symmetric downslanting palpebral fissures,
- coloboma or notching of the lateral part of lower eyelids with medial absence or sparse of the eyelashes and tear ducts defect,
- hypoplasia of the facial bones with micrognathia and retrognathia as well as zygomatic bones. Hypoplasia of the zygomatic bones and mandible can cause significant feeding and respiratory difficulties.
3. Management and Surgical Treatment
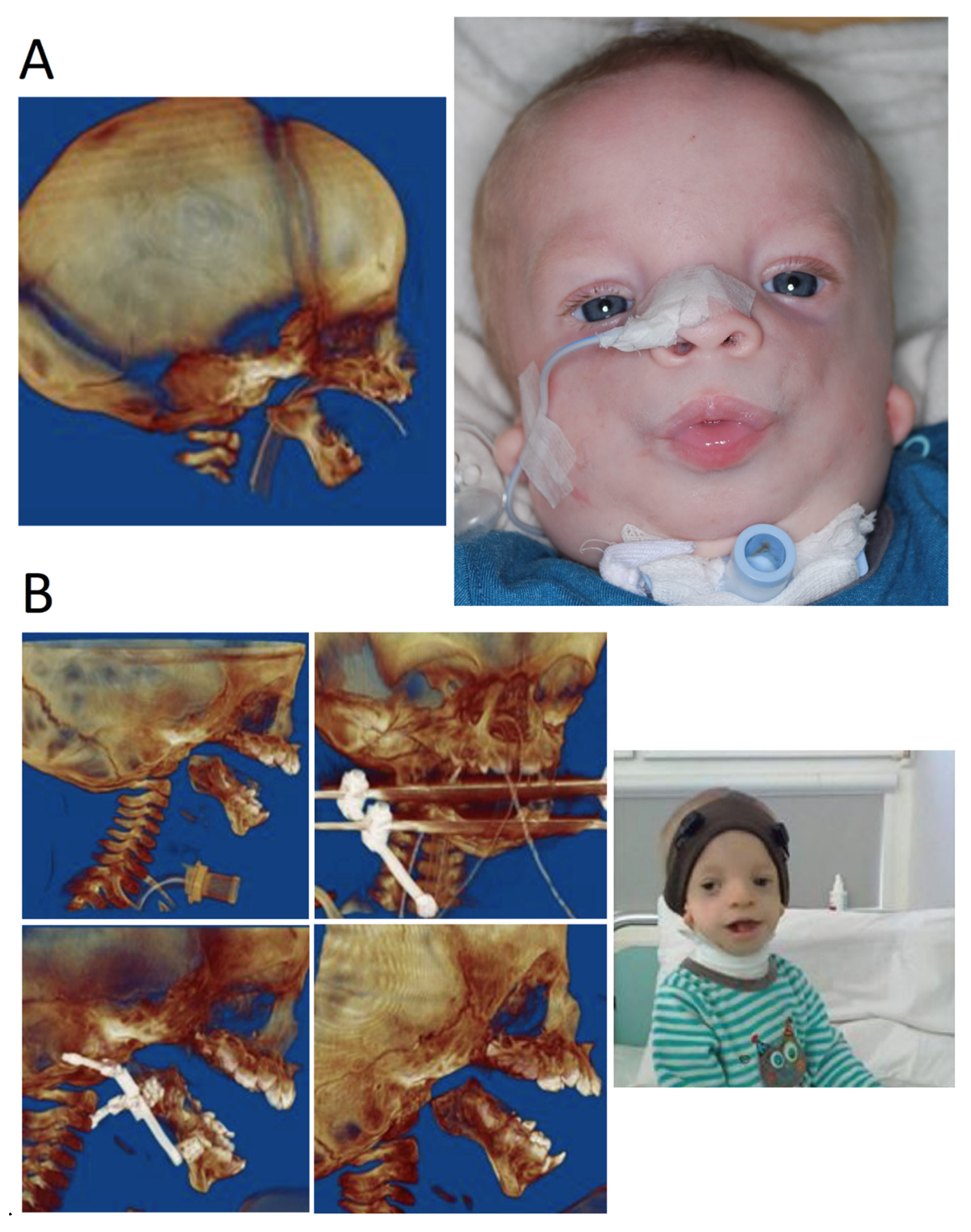
- Birth to age 2 years—facilitation of elementary living functions: breathing, e.g., tracheostomy (Figure 4), feeding and hearing as well as vision and heart.
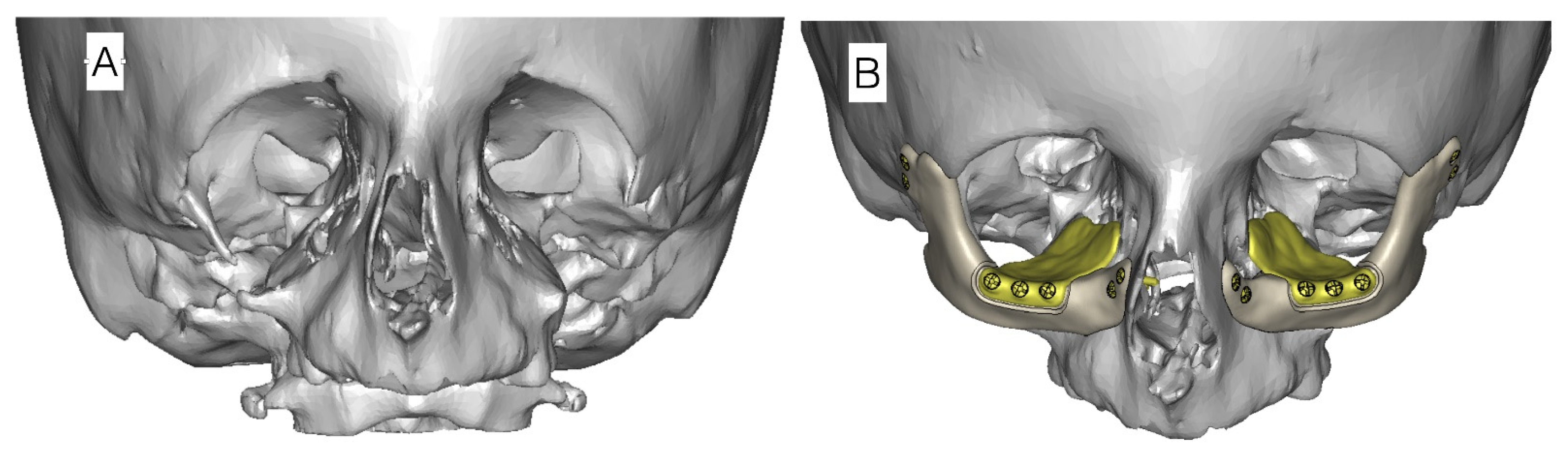
- Age 3 to 12 years—speech therapy, integration into society, bone reconstructions including mandibular (Figure 5), which can prevent progression of defects as well as ophthalmological and orthodontic support. Pre-school children usually receive the same treatment.
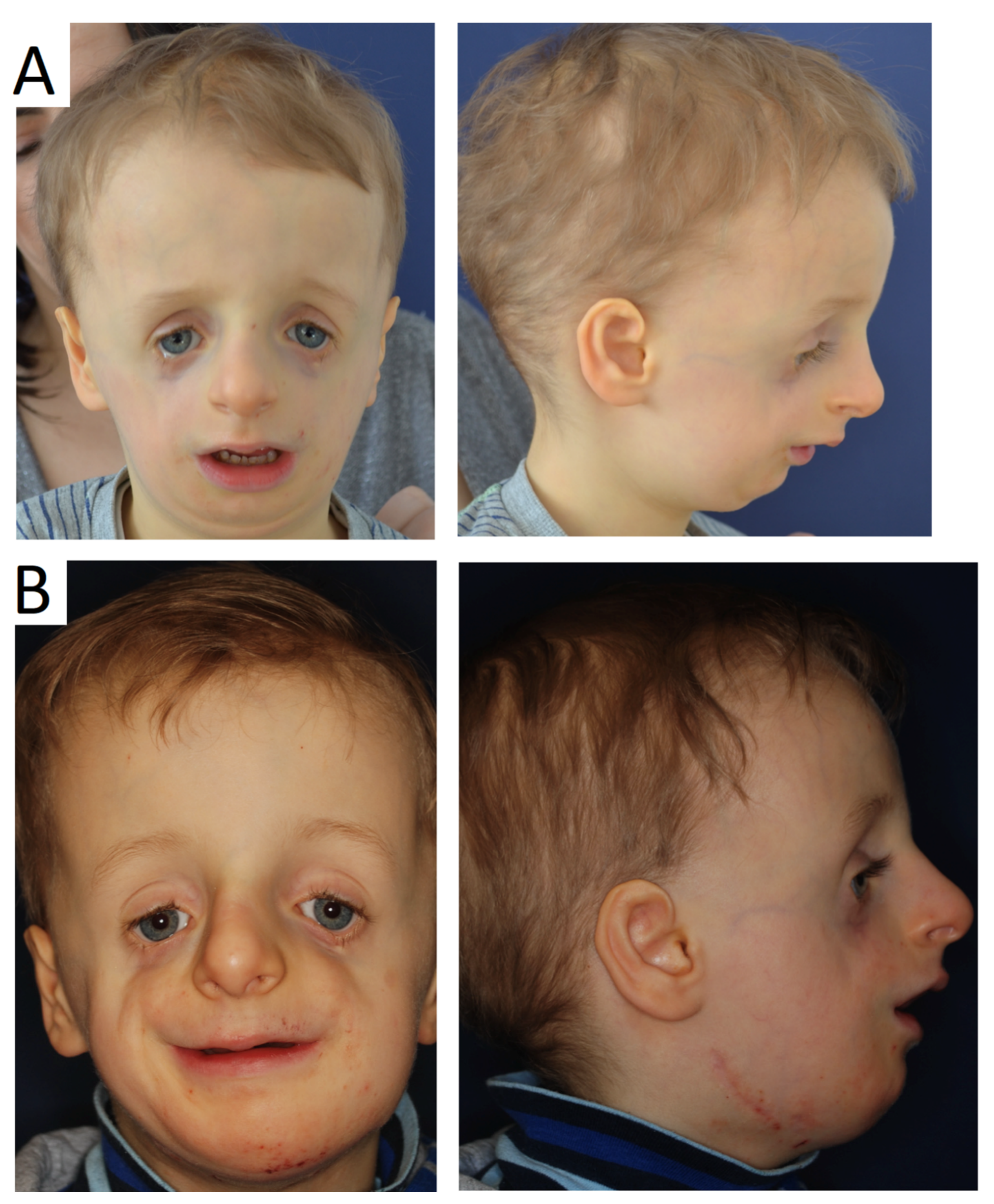
- Age 13 to 18—orthognathic therapies, maxillomandibular and nasal reconstruction as well as integration into society. Multistage reconstructive treatment forming face is possible in this age and makes noticeable improvements (Figure 6). However, if possible, it is recommended to start therapy at an earlier age. The sooner therapy begins gives the higher possibility of achieving optimal results (Figure 7).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, G.A. Note on a congenital defect colobomata of the lower lid. R. Lond. Ophthalmic. Hosp. Rep. 1889, 12, 255. [Google Scholar]
- Collins, E.T. Cases with symmetrical congenital notches in the outer part of each lower lid and defective development of malar bones. Trans. Ophthalmol. Soc. UK 1900, 20, 190. [Google Scholar]
- Franceschetti, A.; Klein, D. The mandibulofacial dysostosis; a new hereditary syndrome. Acta Ophthalmol. 1949, 27, 143–224. [Google Scholar]
- Gorlin, R.J.; Cohen, M.M.; Levin, L.S. Syndromes of the Head and Neck; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Dixon, J.; Trainor, P. Treacher Collins syndrome. Orthod. Craniofacial Res. 2007, 10, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Laplace-Builhé, B.; Mau-Them, F.T.; Richard, E.; Goldenberg, A.; Toler, T.; Guignard, T.; Gatinois, V.; Vincent, M.; Blanchet, C.; et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet. Med. 2020, 22, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauwerse, J.G.; Dixon, J.; Seland, S.; Ruivenkamp, C.A.L.; Van Haeringen, A.; Hoefsloot, L.H.; Peters, D.J.M.; Boers, A.C.-D.; Daumer-Haas, C.; Maiwald, R.; et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2010, 43, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Geneviève, P.; Ostertag, A.; Marlin, S.; Lacombe, D.; Martin-Coignard, D.; Coubes, C.; David, G.; Lyonnet, S.; Vilain, C.; et al. Treacher Collins syndrome: A clinical and molecular study based on a large series of patients. Genet. Med. 2016, 18, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.orpha.net (accessed on 5 August 2021).
- Katsanis, S.H.; Jabs, E.W. Treacher Collins Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2004. [Google Scholar]
- So, R.B.; Gonzales, B.; Henning, D.; Dixon, J.; Dixon, M.J.; Valdez, B.C. Another face of the Treacher Collins syndrome (TCOF1) gene: Identification of additional exons. Gene 2004, 328, 49–57. [Google Scholar] [CrossRef]
- Valdez, B.C.; Henning, D.; So, R.B.; Dixon, J.; Dixon, M.J. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. USA 2004, 101, 10709–10714. [Google Scholar] [CrossRef] [Green Version]
- Isaac, C.; Marsh, K.L.; Paznekas, W.A.; Dixon, J.; Dixon, M.J.; Jabs, E.; Meier, U.T. Characterization of the Nucleolar Gene Product, Treacle, in Treacher Collins Syndrome. Mol. Biol. Cell 2000, 11, 3061–3071. [Google Scholar] [CrossRef] [Green Version]
- Winokur, S.T.; Shiang, R. The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C-terminus. Hum. Mol. Genet. 1998, 7, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Sakai, D.; Trainor, P. Face off against ROS: Tcof1 /Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev. Growth Differ. 2016, 58, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Conte, C.; D’Apice, M.R.; Rinaldi, F.; Gambardella, S.; Sangiuolo, F.; Novelli, G. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome. BMC Med. Genet. 2011, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, S.J.; Zomerdijk, J.C.B.M. Basic Mechanisms in RNA Polymerase I Transcription of the Ribosomal RNA Genes. Subcell. Biochem. 2013, 61, 211–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzanka, M.; Piekiełko-Witkowska, A. The Role of TCOF1 Gene in Health and Disease: Beyond Treacher Collins Syndrome. Int. J. Mol. Sci. 2021, 22, 2482. [Google Scholar] [CrossRef]
- Ciccia, A.; Huang, J.-W.; Izhar, L.; Sowa, M.E.; Harper, J.; Elledge, S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 18631–18636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: www.ncbi.nlm.nih.gov/gene (accessed on 5 August 2021).
- Available online: http://www.ensembl.org (accessed on 5 August 2021).
- Splendore, A.; Fanganiello, R.D.; Masotti, C.; Morganti, L.S.; Passos-Bueno, M.R. TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature. Hum. Mutat. 2005, 25, 429–434. [Google Scholar] [CrossRef]
- Marszałek-Kruk, B.A.; Śmigiel, R.; Sasiadek, M. Novel mutation in the TCOF1 gene in a patient with Treacher Collins syndrome. Pediatr. Polska 2014, 89, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Lu, Y.; Wang, Y.; Zhang, X.; Duan, H.; Cheng, J.; Yuan, H.; Han, D. Identification of a novel TCOF1 mutation in a Chinese family with Treacher Collins syndrome. Exp. Ther. Med. 2018, 16, 2645–2650. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Wang, Y.; Fan, Y.; Du, H.; Luo, N.; Zhang, S.; Chen, X. TCOF1 pathogenic variants identified by Whole-exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J. Rare Dis. 2019, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Xie, M.; Li, J.; Xie, H.; Lu, X. A novel nonsense mutation in the TCOF1 gene in one Chinese newborn with Treacher Collins syndrome. Int. J. Pediatr. Otorhinolaryngol. 2021, 141, 110561. [Google Scholar] [CrossRef]
- Zhang, C.; An, L.; Xue, H.; Hao, S.; Yan, Y.; Zhang, Q.; Jin, X.; Li, Q.; Zhou, B.; Feng, X.; et al. Mutation analysis of TCOF1 gene in Chinese Treacher Collins syndrome patients. J. Clin. Lab. Anal. 2021, 35, e23567. [Google Scholar] [CrossRef] [PubMed]
- Chen-Long, L.; Guo, L.; Li, C.-L.; Shan, J.; Xu, H.-S.; Li, J.-Y.; Sun, S.; Hao, S.-J.; Jing, S.; Chai, G.; et al. Mutation screening of Chinese Treacher Collins syndrome patients identified novel TCOF1 mutations. Mol. Genet. Genom. 2018, 293, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Oldridge, M.; Archer, C.; O’Rourke, A.; McParland, J.; Brekelmans, R.; Seller, A.; Lester, T. Gross deletions in TCOF1 are a cause of Treacher–Collins–Franceschetti syndrome. Eur. J. Hum. Genet. 2012, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Beygo, J.; Buiting, K.; Seland, S.; Lüdecke, H.-J.; Hehr, U.; Lich, C.; Prager, B.; Lohmann, D.; Wieczorek, D. First Report of a Single Exon Deletion in TCOF1 Causing Treacher Collins Syndrome. Mol. Syndr. 2012, 2, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lin, P.; Pang, J.; Jia, Z.; Peng, Y.; Xi, H.; Wu, L.; Li, Z.; Wang, H. Identification of a novel gross deletion of TCOF1 in a Chinese prenatal case with Treacher Collins syndrome. Mol. Genet. Genom. Med. 2020, 8, e1313. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Tripuwabhrut, K.; Intachai, W.; Carlson, B.M.; Quarto, N.; Ngamphiw, C.; Tongsima, S.; Sonsuwan, N. Treacher Collins syndrome: A novel TCOF1 mutation and monopodial stapes. Clin. Otolaryngol. 2020, 45, 695–702. [Google Scholar] [CrossRef]
- Marszałek-Kruk, B.A.; Wojcicki, P.; Śmigiel, R.; Trzeciak, W.H. Novel insertion in exon 5 of the TCOF1 gene in twin sisters with Treacher Collins syndrome. J. Appl. Genet. 2012, 53, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Teber, A.; Gillessen-Kaesbach, G.; Fischer, S.; Böhringer, S.; Albrecht, B.; Albert, A.; Arslan-Kirchner, M.; Haan, E.; Hagedorn-Greiwe, M.; Hammans, C.; et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur. J. Hum. Genet. 2004, 12, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Splendore, A.; Silva, E.O.; Alonso, L.G.; Richieri-Costa, A.; Alonso, N.; Rosa, A.; Carakushanky, G.; Cavalcanti, D.P.; Brunoni, D.; Passos-Bueno, M.R. High mutation detection rate in TCOF1 among Treacher Collins syndrome patients reveals clustering of mutations and 16 novel pathogenic changes. Hum. Mutat. 2000, 16, 315–322. [Google Scholar] [CrossRef]
- Posnick, J.C.; Ruiz, R.L. Treacher Collins syndrome: Current evaluation, treatment, and future directions. Cleft Palate Craniofac. J. 2000, 37, 434. [Google Scholar] [CrossRef]
- Wójcicki, P.; Marszałek-Kruk, B. Genetic Factors and Treatment Principles of Treacher Collins Syndrome. Dent. Med. Probl. 2005, 42, 619–626. [Google Scholar]
- Jackson, I.T. Reconstruction of the Lower Eyelid Defect in Treacher Collins Syndrome. Plast. Reconstr. Surg. 1981, 67, 365–367. [Google Scholar] [CrossRef]
- Posnick, J.C. Treacher Collins syndrome: Perspectives in evaluation and treatment. J. Oral Maxillofac. Surg. 1997, 55, 1120–1133. [Google Scholar] [CrossRef]
- Farkas, L.G.; Posnick, J.C. Detailed Morphometry of the Nose in Patients with Treacher Collins Syndrome. Ann. Plast. Surg. 1989, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Gene | |||||||
|---|---|---|---|---|---|---|---|---|
| TCS Subtype | OMIM | Name | OMIM | Inheritance | ID | Chromosome Locus | Frequency of TCS Pathogenic Variant [6,7,8] | Product of Gene |
| TCS1 | 154500 | TCOF1 | 606847 | AD | 6949 | 5q32-q33 | 86% | Treacle protein |
| TCS2 | 613717 | POLR1D | 613715 | AD, AR | 51082 | 13q12.2 | 6% | DNA-directed RNA polymerases I and III subunit RPAC2 |
| TCS3 | 248390 | POLR1C | 610060 | AR | 9533 | 6p21.1 | 1.2% | DNA-directed RNA polymerases I and III subunit RPAC1 |
| TCS4 | 618939 | POLR1B | 602000 | AD | 84172 | 2q14.1 | 1.3% | DNA-directed RNA polymerase I subunit RPA2 |
| Classic Feature | Symptom, Feature | Occurrence in Affected Individuals |
|---|---|---|
| Very frequent | Downslanting palpebral fissures | 89–100% |
| Malar hypoplasia/hypoplasia of zygomatic complex | 81–97% | |
| Conductive hearing loss | 83–92% | |
| Mandibular hypoplasia/micrognathia | 78–91% | |
| Frequent | Atresia of external ear canal | 68–71% |
| Microtia | 10–77% | |
| Coloboma (notching) of the lower lid | 54–69% | |
| Delayed speech development | 57–63% | |
| Asymmetry | 52% | |
| Preauricular hair displacement | 24–49% | |
| Rare | Nasogastric tube or gastrostomy in neonates | 28% |
| Cleft palate | 21–33% | |
| Intubation or tracheostomy in neonates | 12–18% | |
| Choanal stenosis/atresia | 13–25% | |
| Cardiac malformation | 11% | |
| Very rare | Rachis malformation | 7% |
| Renal malformation | 4% | |
| Microcephaly | 3% | |
| Intellectual disability Delayed motor development | 1.7–10% | |
| Limb anomaly | 1.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marszałek-Kruk, B.A.; Wójcicki, P.; Dowgierd, K.; Śmigiel, R. Treacher Collins Syndrome: Genetics, Clinical Features and Management. Genes 2021, 12, 1392. https://doi.org/10.3390/genes12091392
Marszałek-Kruk BA, Wójcicki P, Dowgierd K, Śmigiel R. Treacher Collins Syndrome: Genetics, Clinical Features and Management. Genes. 2021; 12(9):1392. https://doi.org/10.3390/genes12091392
Chicago/Turabian StyleMarszałek-Kruk, Bożena Anna, Piotr Wójcicki, Krzysztof Dowgierd, and Robert Śmigiel. 2021. "Treacher Collins Syndrome: Genetics, Clinical Features and Management" Genes 12, no. 9: 1392. https://doi.org/10.3390/genes12091392
APA StyleMarszałek-Kruk, B. A., Wójcicki, P., Dowgierd, K., & Śmigiel, R. (2021). Treacher Collins Syndrome: Genetics, Clinical Features and Management. Genes, 12(9), 1392. https://doi.org/10.3390/genes12091392