
Differential Expression of Long Non-Coding RNA (lncRNA) in Mediterranean Mussel (Mytilus galloprovincialis) Hemocytes under Immune Stimuli


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
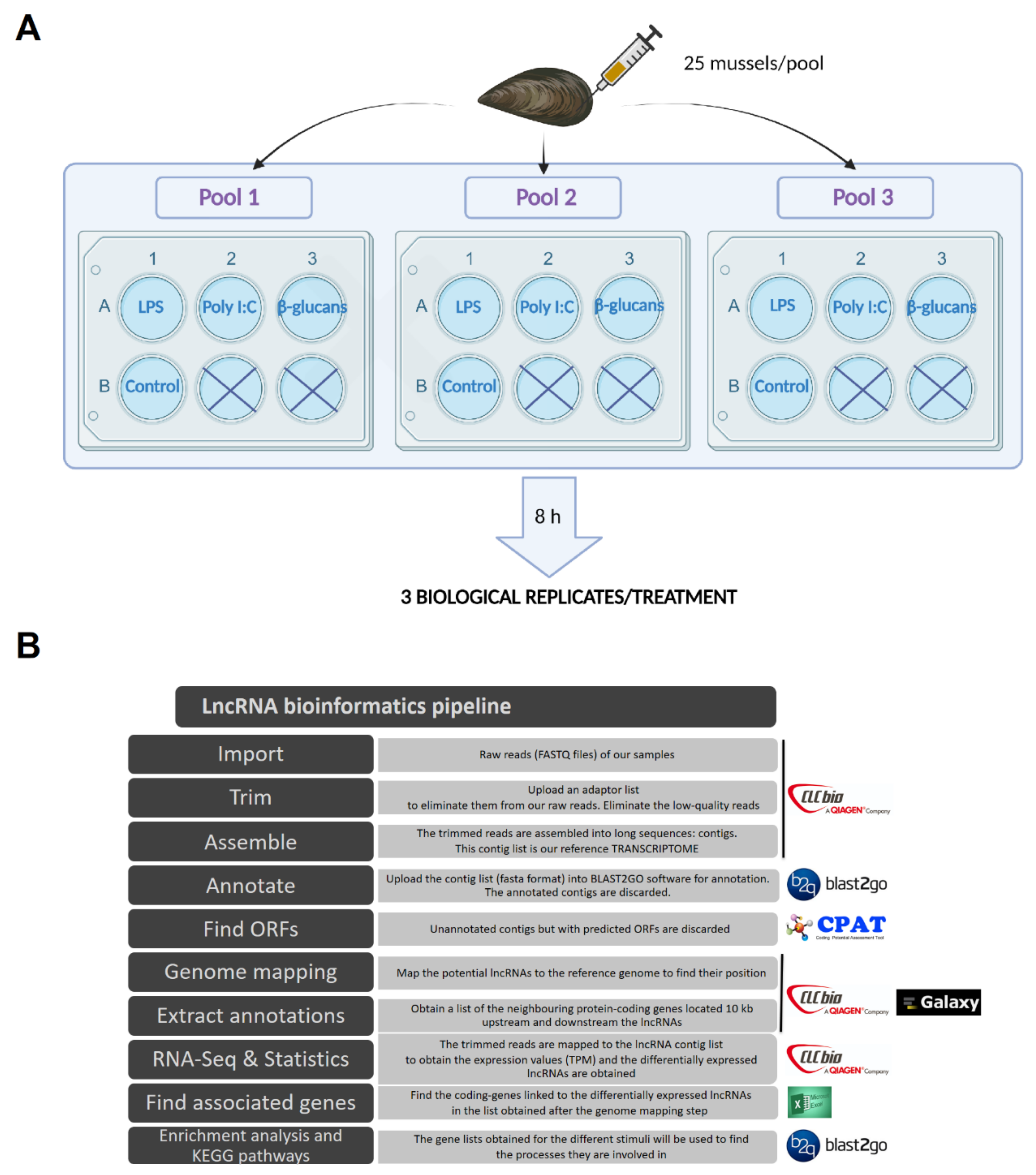
2.2. Experimental Design, RNA Isolation, and Illumina Sequencing
2.3. Trimming, Sequence Assembly, and lncRNA Mining
2.4. Genome Mapping and Identification of lncRNA-Neighboring Coding Genes
2.5. RNA-Seq and Differential Expression Analyses
2.6. Gene Ontology (GO) Enrichment Analysis and KEGG Pathways
2.7. Correlation Analyses between lncRNAs and Coding Genes
2.8. Quantitative PCR (qPCR) Validation of Differentially Expressed lncRNAs
3. Results
3.1. Assembly, Annotation, and lncRNA Mining
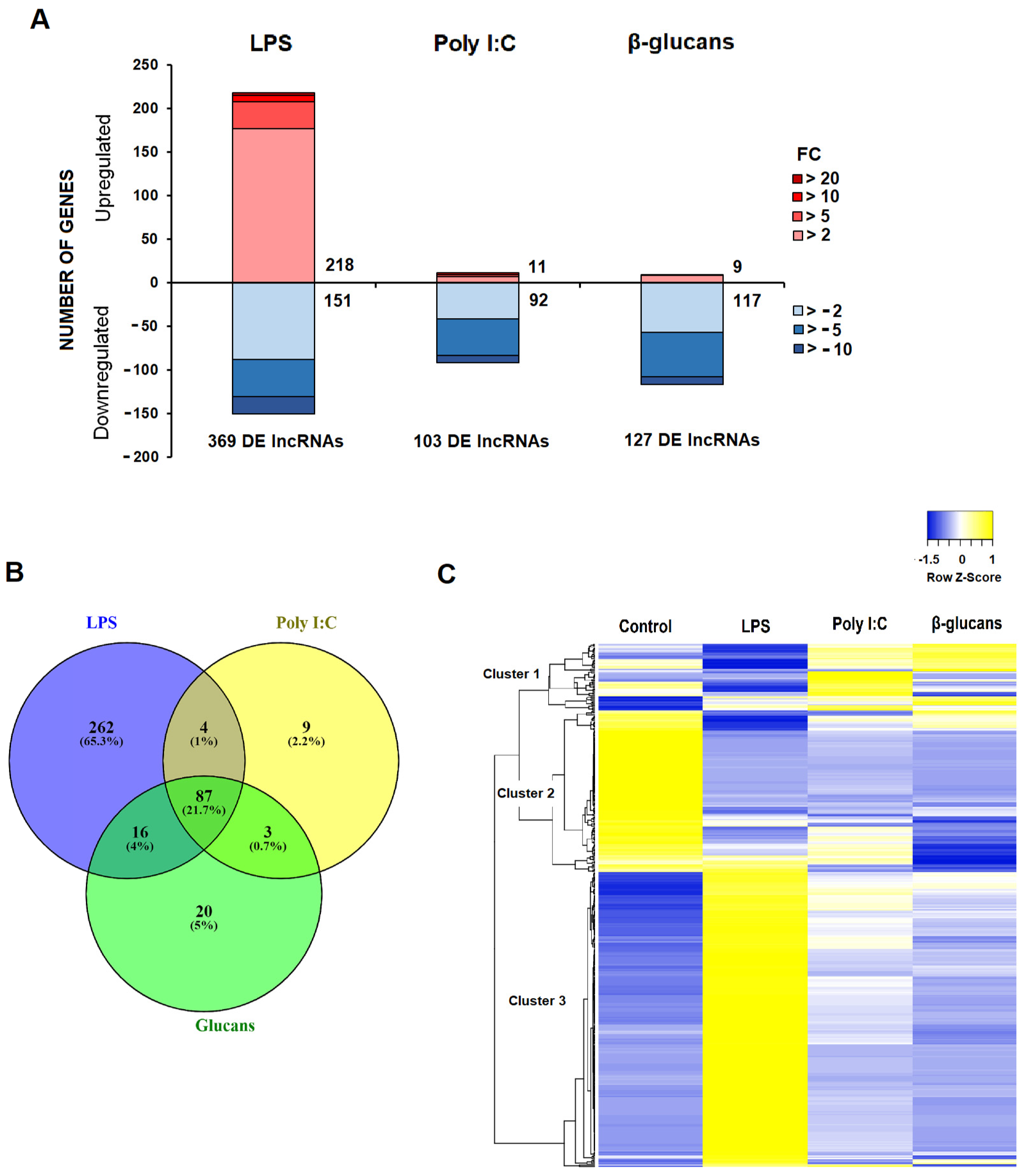
3.2. Differential Expression of lncRNAs after Stimulation of Hemocytes with PAMPs
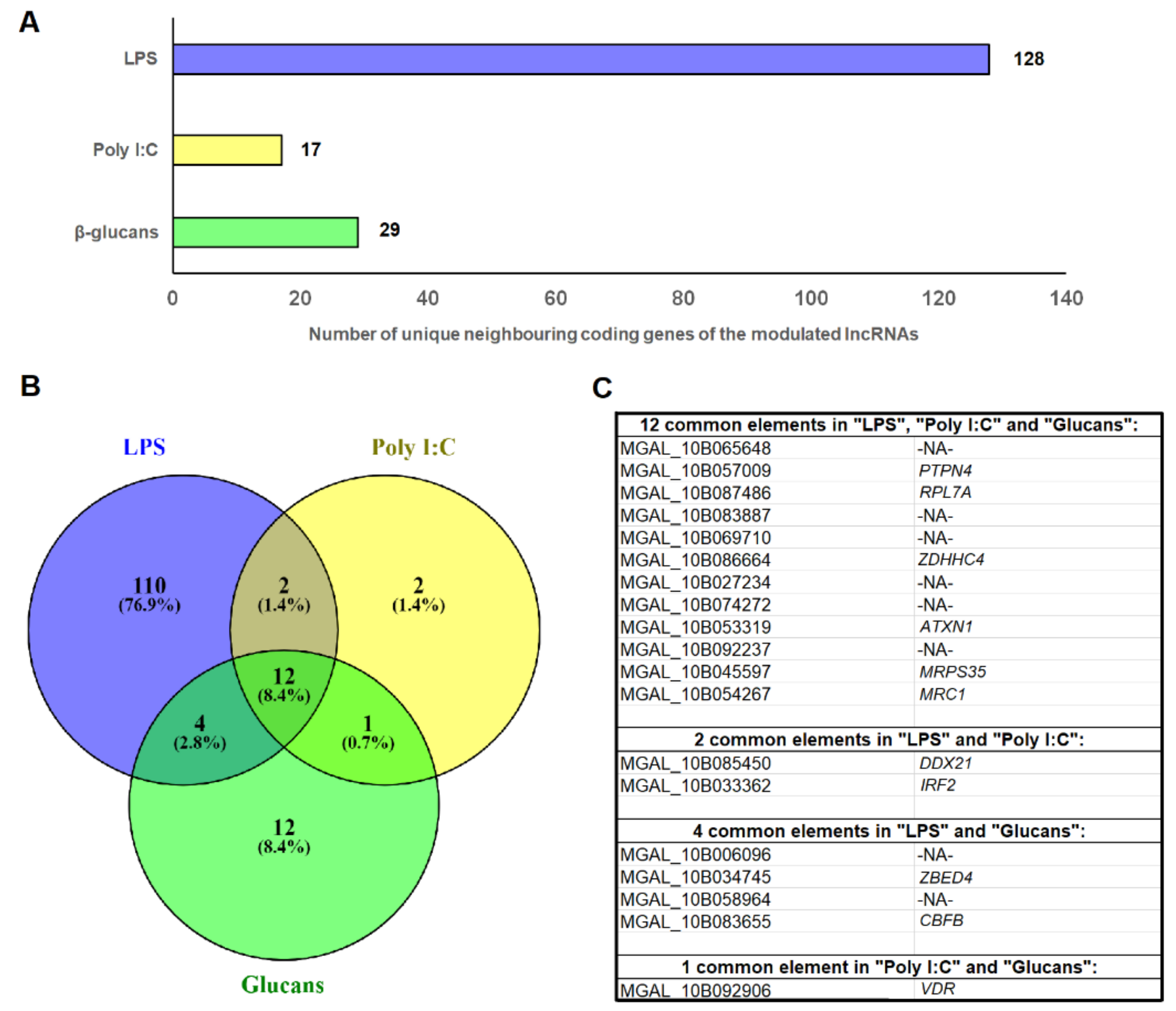
3.3. Identification of the DE lncRNA-Neighboring Coding Genes
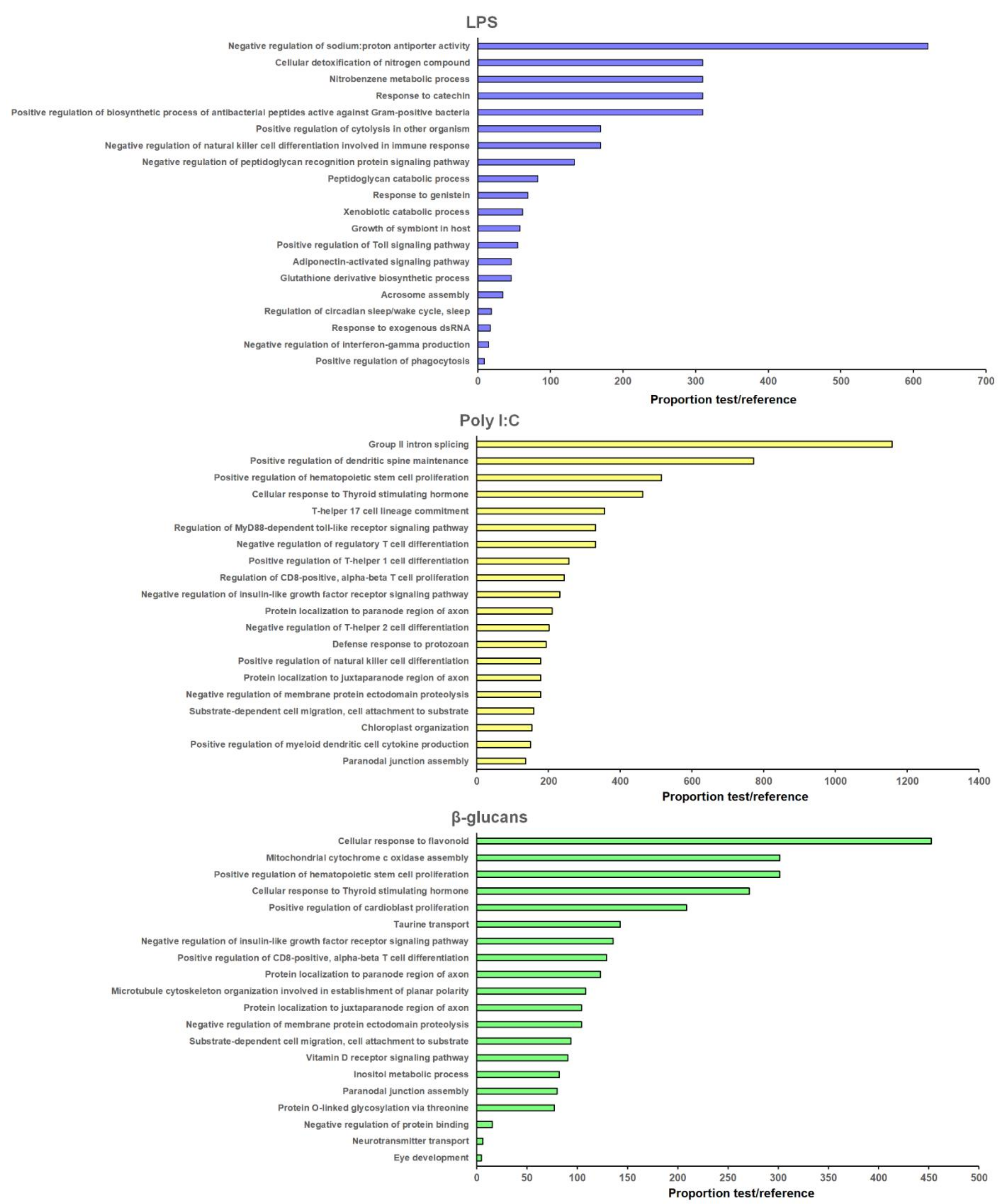
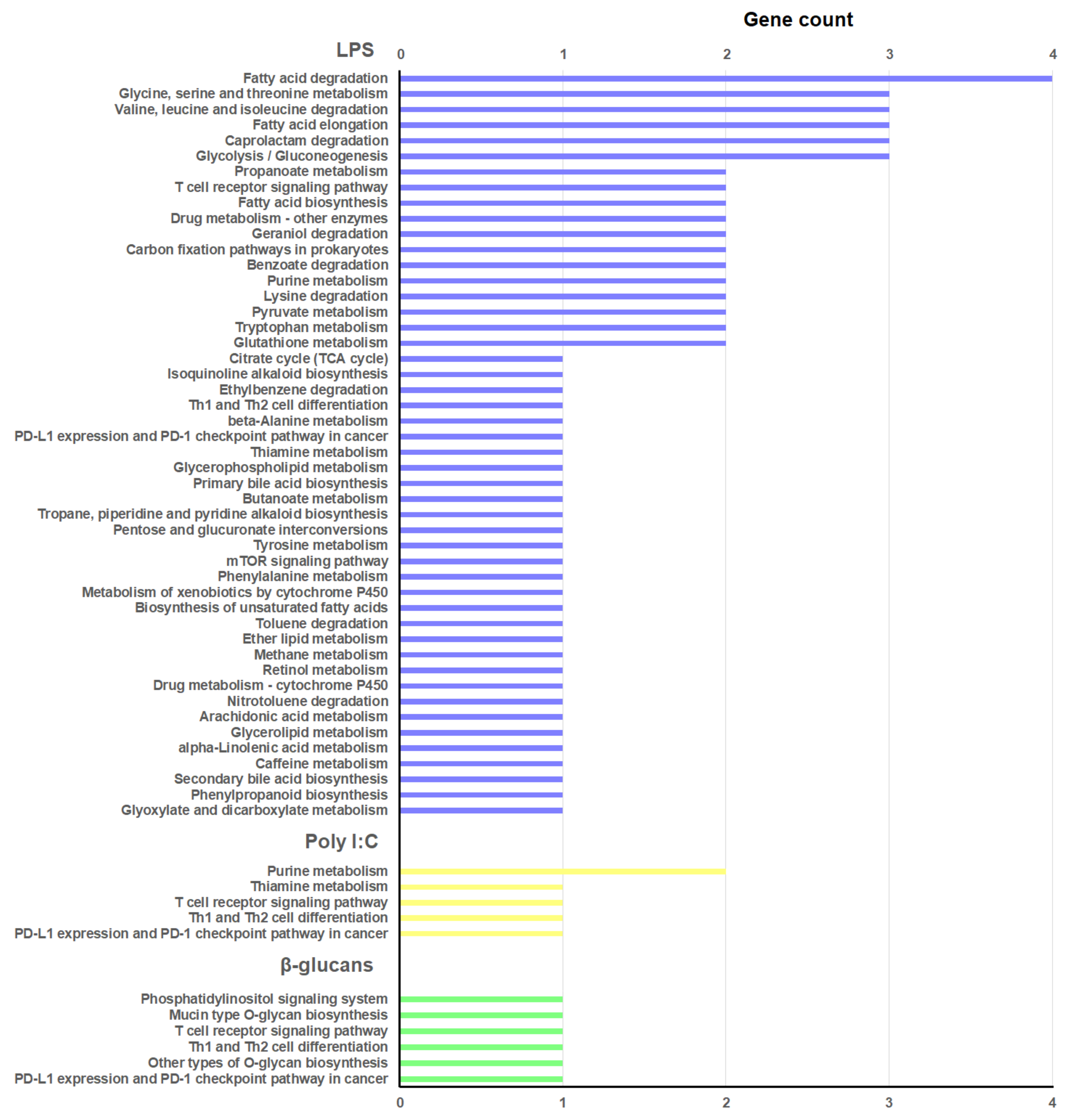
3.4. GO Enrichment and KEGG Pathway Analyses of the lncRNA-Neighboring Coding Genes
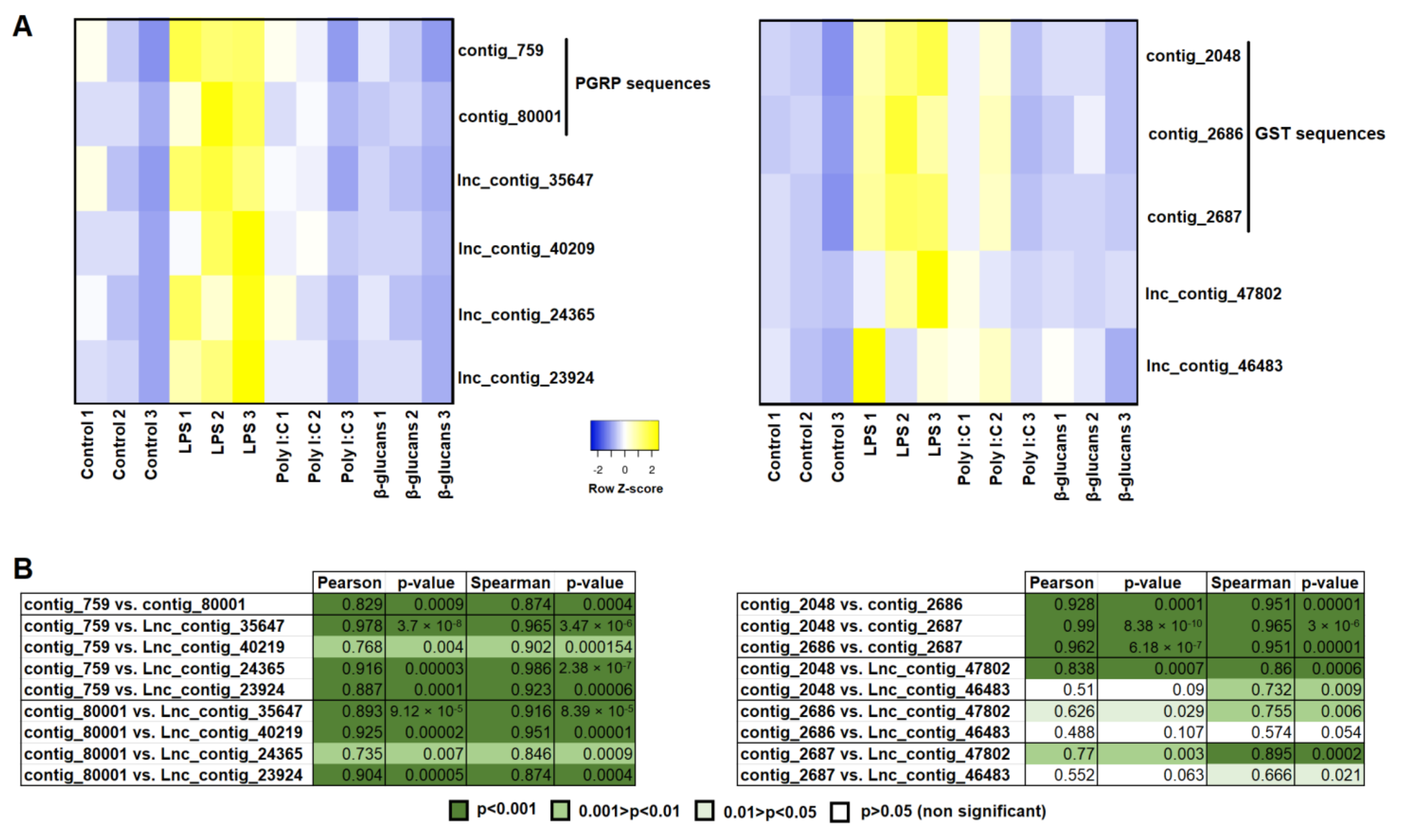
3.5. Correlation Analysis between Modulated Genes and lncRNAs
3.6. Validation of lncRNA Expression Profiles by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. Food and Agriculture Organization (2004–2020). FAO Fisheries Division; Cultured Aquatic Species Information Programme: Mytilus galloprovincialis. Available online: http://www.fao.org/fishery/culturedspecies/Mytilus_galloprovincialis/en (accessed on 8 September 2021).
- Kurelec, B.; Pivčević, B. Evidence for a multixenobiotic resistance mechanism in the mussel Mytilus galloprovincialis. Aquat. Toxicol. 1991, 19, 291–301. [Google Scholar] [CrossRef]
- Gestal, C.; Roch, P.; Renault, T.; Pallavicini, A.; Paillard, C.; Novoa, B.; Oubella, R.; Venier, P.; Figueras, A. Study of diseases and the immune system of bivalves using molecular biology and genomics. Rev. Fish. Sci. Aquac. 2008, 16, 133–156. [Google Scholar] [CrossRef] [Green Version]
- Domeneghetti, S.; Varotto, L.; Civettini, M.; Rosani, U.; Stauder, M.; Pretto, T.; Pezzati, E.; Arcangeli, G.; Turolla, E.; Pallavicini, A.; et al. Mortality occurrence and pathogen detection in Crassostrea gigas and Mytilus galloprovincialis close-growing in shallow waters (Goro lagoon, Italy). Fish. Shellfish Immunol. 2014, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Costa, M.M.; Forn-Cuni, G.; Balseiro, P.; Chamorro, R.; Dios, S.; Figueras, A.; Novoa, B. Occurrence, seasonality and infectivity of Vibrio strains in natural populations of mussels Mytilus galloprovincialis. Dis. Aquat. Organ. 2014, 108, 149–163. [Google Scholar] [CrossRef]
- Mitta, G.; Vandenbulcke, F.; Roch, P. Original involvement of antimicrobial peptides in mussel innate immunity. FEBS Lett. 2000, 486, 85–190. [Google Scholar] [CrossRef] [Green Version]
- Pallavicini, A.; Costa, M.M.; Gestal, C.; Dreos, R.; Figueras, A.; Venier, P.; Novoa, B. High sequence variability of myticin transcripts in hemocytes of immune-stimulated mussels suggests ancient host-pathogen interactions. Dev. Comp. Immunol. 2008, 32, 213–226. [Google Scholar] [CrossRef]
- Costa, M.M.; Dios, S.; Alonso-Gutierrez, J.; Romero, A.; Novoa, B.; Figueras, A. Evidence of high individual diversity on myticin C in mussel (Mytilus galloprovincialis). Dev. Comp. Immunol. 2009, 33, 162–170. [Google Scholar] [CrossRef]
- Novoa, B.; Romero, A.; Álvarez, Á.L.; Moreira, R.; Pereiro, P.; Costa, M.M.; Dios, S.; Estepa, A.; Parra, F.; Figueras, A. Antiviral activity of myticin C peptide from mussel: An ancient defense against herpesviruses. J. Virol. 2016, 90, 7692–7702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdol, M.; Manfrin, C.; De Moro, G.; Figueras, A.; Novoa, B.; Venier, P.; Pallavicini, A. The C1q domain containing proteins of the Mediterranean mussel Mytilus galloprovincialis: A widespread and diverse family of immune-related molecules. Dev. Comp. Immunol. 2011, 35, 635–643. [Google Scholar] [CrossRef]
- Romero, A.; Dios, S.; Poisa-Beiro, L.; Costa, M.M.; Posada, D.; Figueras, A.; Novoa, B. Individual sequence variability and functional activities of fibrinogen-related proteins (FREPs) in the Mediterranean mussel (Mytilus galloprovincialis) suggest ancient and complex immune recognition models in invertebrates. Dev. Comp. Immunol. 2011, 35, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Toubiana, M.; Gerdol, M.; Rosani, U.; Pallavicini, A.; Venier, P.; Roch, P. Toll-like receptors and MyD88 adaptors in Mytilus: Complete cds and gene expression levels. Dev. Comp. Immunol. 2013, 40, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Varotto, L.; Gerdol, M.; Pallavicini, A.; Venier, P. IL-17 signaling components in bivalves: Comparative sequence analysis and involvement in the immune responses. Dev. Comp. Immunol. 2015, 52, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdol, M.; Venier, P. An updated molecular basis for mussel immunity. Fish Shellfish. Immunol. 2015, 46, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueguen, Y.; Cadoret, J.P.; Flament, D.; Barreau-Roumiguière, C.; Girardot, A.L.; Garnier, J.; Hoareau, A.; Bachère, E.; Escoubas, J.M. Immune gene discovery by expressed sequence tags generated from hemocytes of the bacteria-challenged oyster, Crassostrea gigas. Gene 2003, 303, 139–145. [Google Scholar] [CrossRef]
- De Lorgeril, J.; Zenagui, R.; Rosa, R.D.; Piquemal, D.; Bachère, E. Whole transcriptome profiling of successful immune response to Vibrio infections in the oyster Crassostrea gigas by digital gene expression analysis. PLoS ONE 2011, 6, e23142. [Google Scholar] [CrossRef] [Green Version]
- Green, T.J.; Montagnani, C. Poly I:C induces a protective antiviral immune response in the Pacific oyster (Crassostrea gigas) against subsequent challenge with Ostreid herpesvirus (OsHV-1 μvar). Fish. Shellfish Immunol. 2013, 35, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Zhang, L.; Huang, B.; Li, L.; Zhang, G. Comparative analysis of oyster (Crassostrea gigas) immune responses under challenge by different Vibrio strains and conditions. Molluscan Res. 2015, 35, 1–11. [Google Scholar] [CrossRef]
- Green, T.J.; Vergnes, A.; Montagnani, C.; de Lorgeril, J. Distinct immune responses of juvenile and adult oysters (Crassostrea gigas) to viral and bacterial infections. Vet. Res. 2016, 47, 72. [Google Scholar] [CrossRef] [Green Version]
- Lafont, M.; Vergnes, A.; Vidal-Dupiol, J.; de Lorgeril, J.; Gueguen, Y.; Haffner, P.; Petton, B.; Chaparro, C.; Barrachina, C.; Destoumieux-Garzon, D.; et al. A sustained immune response supports long-term antiviral immune priming in in the Pacific Oyster, Crassostrea Gigas. Mbio 2020, 11, e02777-19. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.M.; Prado-Alvarez, M.; Gestal, C.; Li, H.; Roch, P.; Novoa, B.; Figueras, A. Functional and molecular immune response of Mediterranean mussel (Mytilus galloprovincialis) haemocytes against pathogen-associated molecular patterns and bacteria. Fish. Shellfish Immunol. 2009, 26, 515–523. [Google Scholar] [CrossRef]
- Venier, P.; Varotto, L.; Rosani, U.; Millino, C.; Celegato, B.; Bernante, F.; Lanfranchi, G.; Novoa, B.; Roch, P.; Figueras, A.; et al. Insights into the innate immunity of the Mediterranean mussel Mytilus Galloprovincialis. BMC Genom. 2011, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Balseiro, P.; Moreira, R.; Chamorro, R.; Figueras, A.; Novoa, B. Immune responses during the larval stages of Mytilus galloprovincialis: Metamorphosis alters immunocompetence, body shape and behavior. Fish. Shellfish Immunol. 2013, 35, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Rey-Campos, M.; Moreira, R.; Valenzuela-Muñoz, V.; Gallardo-Escárate, C.; Novoa, B.; Figueras, A. High individual variability in the transcriptomic response of Mediterranean mussels to Vibrio reveals the involvement of myticins in tissue injury. Sci. Rep. 2019, 9, 3569. [Google Scholar] [CrossRef] [PubMed]
- Rey-Campos, M.; Moreira, R.; Gerdol, M.; Pallavicini, A.; Novoa, B.; Figueras, A. Immune Tolerance in Mytilus galloprovincialis hemocytes after repeated contact with Vibrio splendidus. Front. Immunol. 2019, 10, 1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, R.; Romero, A.; Rey-Campos, M.; Pereiro, P.; Rosani, U.; Novoa, B.; Figueras, A. Stimulation of Mytilus galloprovincialis hemocytes with different immune challenges induces differential transcriptomic, miRNomic, and functional responses. Front. Immunol. 2020, 11, 606102. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Mu, C.; Wang, R.; Li, T.; Li, Y.; Tian, M.; Jiao, W.; Huang, X.; Zhang, L.; Hu, X.; Wang, S.; et al. Long non-coding RNAs (lncRNAs) of sea cucumber: Large-scale prediction, expression profiling, non-coding network construction, and lncRNA-microRNA-Gene interaction analysis of lncRNAs in Apostichopus japonicus and Holothuria glaberrima during LPS challenge and radial organ complex regeneration. Mar. Biotechnol. 2016, 18, 485–499. [Google Scholar] [CrossRef]
- Sun, W.; Feng, J. Differential lncRNA expression profiles reveal the potential roles of lncRNAs in antiviral immune response of Crassostrea gigas. Fish. Shellfish Immunol. 2018, 81, 233–241. [Google Scholar] [CrossRef]
- Ali, A.; Abd El Halim, H.M. Re-thinking adaptive immunity in the beetles: Evolutionary and functional trajectories of lncRNAs. Genomics 2020, 112, 1425–1436. [Google Scholar] [CrossRef]
- Pang, K.C.; Dinger, M.E.; Mercer, T.R.; Malquori, L.; Grimmond, S.M.; Chen, W.; Mattick, J.S. Genome-wide identification of long noncoding RNAs in CD8+ T cells. J. Immunol. 2009, 182, 7738–7748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. Mbio 2010, 1, e00206–e00210. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela-Muñoz, V.; Pereiro, P.; Álvarez-Rodríguez, M.; Gallardo-Escárate, C.; Figueras, A.; Novoa, B. Comparative modulation of lncRNAs in wild-type and rag1-heterozygous mutant zebrafish exposed to immune challenge with spring viraemia of carp virus (SVCV). Sci. Rep. 2019, 9, 14174. [Google Scholar] [CrossRef]
- Pereiro, P.; Lama, R.; Moreira, R.; Valenzuela-Muñoz, V.; Gallardo-Escárate, C.; Novoa, B.; Figueras, A. Potential involvement of lncRNAs in the modulation of the transcriptome response to nodavirus challenge in European sea bass (Dicentrarchus labrax L.). Biology 2020, 9, 165. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Hasan, A.M.; Farzaneh, F.; Williams, G.T. Inhibition of human T cell proliferation by mammalian target of rapamycin (mTOR) antagonists requires noncoding RNA growth-arrest-specific transcript 5 (GAS5). Mol. Pharmacol. 2010, 78, 19–28. [Google Scholar] [CrossRef]
- Rapicavoli, N.A.; Qu, K.; Zhang, J.; Mikhail, M.; Laberge, R.M.; Chang, H.Y.; Gingera, T. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife 2013, 2, e00762. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Pang, W.; Wang, Y.; Xiong, Y.; Xu, R.; Wu, W.; Zhao, C.; Yang, G. Knockdown of PU.1 mRNA and AS lncRNA regulates expression of immune-related genes in zebrafish Danio rerio. Dev. Comp. Immunol. 2014, 44, 315–319. [Google Scholar] [CrossRef]
- Huang, X.D.; Dai, J.G.; Lin, K.T.; Liu, M.; Ruan, H.T.; Zhang, H.; Liu, W.G.; He, M.X.; Zhao, M. Regulation of IL-17 by lncRNA of IRF-2 in the pearl oyster. Fish. Shellfish Immunol. 2018, 81, 108–112. [Google Scholar] [CrossRef]
- Valanne, S.; Salminen, T.S.; Järvelä-Stölting, M.; Vesala, L.; Rämet, M. Immune-inducible non-coding RNA molecule lincRNA-IBIN connects immunity and metabolism in Drosophila melanogaster. PLoS Pathog. 2019, 15, e1007504. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef] [Green Version]
- Hongkuan, Z.; Karsoon, T.; Shengkang, L.; Hongyu, M.; Huaiping, Z. The functional roles of the non-coding RNAs in molluscs. Gene 2021, 768, 145300. [Google Scholar] [CrossRef]
- Saco, A.; Rey-Campos, M.; Novoa, B.; Figueras, A. Transcriptomic response of mussel gills after a Vibrio splendidus infection demonstrates their role in the immune response. Front. Immunol. 2020, 11, 615580. [Google Scholar] [CrossRef] [PubMed]
- Tarifeño-Saldivia, E.; Valenzuela-Miranda, D.; Gallardo-Escárate, C. In the shadow: The emerging role of long non-coding RNAs in the immune response of Atlantic salmon. Dev. Comp. Immunol. 2017, 73, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Gerdol, M.; Moreira, R.; Cruz, F.; Gómez-Garrido, J.; Vlasova, A.; Rosani, U.; Venier, P.; Naranjo-Ortiz, M.A.; Murgarella, M.; Greco, S.; et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 2020, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Moreira, R.; Pereiro, P.; Costa, M.; Figueras, A.; Novoa, B. Evaluation of reference genes of Mytilus galloprovincialis and Ruditapes philippinarum infected with three bacteria strains for gene expression analysis. Aquat. Living Resour. 2014, 27, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zhang, C.; Jiang, F.; Wang, Y.; Xu, Z.; Wang, C. Bioinformatics analysis of hemocyte miRNAs of scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish. Shellfish Immunol. 2014, 37, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zheng, Z.; Du, X.; Wang, Q.; Huang, R.; Deng, Y.; Shi, S.; Zhao, X. Identification and characterization of MicroRNAs in pearl oyster Pinctada martensii by Solexa deep sequencing. Mar. Biotechnol. 2014, 16, 54–62. [Google Scholar] [CrossRef]
- Martín-Gómez, L.; Villalba, A.; Kerkhoven, R.H.; Abollo, E. Role of microRNAs in the immunity process of the flat oyster Ostrea edulis against bonamiosis. Infect. Genet. Evol 2014, 27, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, L.; Song, L.; Liu, R.; Zhang, H.; Huang, M.; Chen, H. The identification and characteristics of immune-related microRNAs in haemocytes of oyster Crassostrea gigas. PLoS ONE 2014, 9, e88397. [Google Scholar] [CrossRef]
- Kenny, N.J.; Namigai, E.K.O.; Marlétaz, F.; Hui, J.H.L.; Shimeld, S.M. Draft genome assemblies and predicted microRNA complements of the intertidal lophotrochozoans Patella vulgata (Mollusca, Patellogastropoda) and Spirobranchus (Pomatoceros) lamarcki (Annelida, Serpulida). Mar. Genom. 2015, 24, 139–146. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiao, Y.; Du, X.D.; Tian, Q.L.; Wang, Q.H.; Huang, R.L.; Deng, Y.W. Computational prediction of candidate miRNAs and their potential functions in biomineralization in pearl oyster Pinctada martensii. Saudi J. Biol. Sci. 2016, 23, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Luo, X.; Huang, M.; Liu, G.; You, W.; Ke, C. Identification and characteristics of muscle growth-related microRNA in the Pacific abalone, Haliotis discus hannai. BMC Genom. 2018, 19, 915. [Google Scholar] [CrossRef]
- Yu, D.; Wu, H.; Peng, X.; Ji, C.; Zhang, X.; Song, J.; Qu, J. Profiling of microRNAs and mRNAs in marine mussel Mytilus galloprovincialis. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2020, 230, 108697. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, X.; Li, Q. Genome-wide identification and characterization of long intergenic noncoding RNAs and their potential association with larval development in the Pacific oyster. Sci. Rep. 2016, 6, 20796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Détrée, C.; Núñez-Acuña, G.; Tapia, F.; Gallardo-Escárate, C. Long non-coding RNAs are associated with spatiotemporal gene expression profiles in the marine gastropod Tegula atra. Mar. Genom. 2017, 33, 39–45. [Google Scholar] [CrossRef]
- Huang, J.; Luo, X.; Zeng, L.; Huang, Z.; Huang, M.; You, W.; Ke, C. Expression profiling of lncRNAs and mRNAs reveals regulation of muscle growth in the Pacific abalone, Haliotis discus hannai. Sci. Rep. 2018, 8, 16839. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Li, Q.; Yu, H.; Kong, L.; Du, S. Transcriptional profiling of long non-coding RNAs in mantle of Crassostrea gigas and their association with shell pigmentation. Sci. Rep. 2018, 8, 1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Xiong, X.; Zhang, J.; Lv, S.; Jiao, Y.; Deng, Y. The global effects of PmRunt co-located and co-expressed with a lincRNA lncRunt in pearl oyster Pinctada fucata martensii. Fish. Shellfish Immunol. 2019, 91, 209–215. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, W.; Xu, J.; Xie, B.; Yang, M.; Huang, H.; Li, H.; Wang, Q. LncMSEN1, a mantle-specific LncRNA participating in nacre formation and response to polyI:C stimulation in pearl oyster Pinctada fucata martensii. Fish. Shellfish Immunol. 2020, 96, 330–335. [Google Scholar] [CrossRef]
- Jiang, C.; Li, Y.; Zhao, Z.; Lu, J.; Chen, H.; Ding, N.; Wang, G.; Xu, J.; Li, X. Identifying and functionally characterizing tissue-specific and ubiquitously expressed human lncRNAs. Oncotarget 2016, 7, 7120–7133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.B.T.; Ulitsky, I. The functions of long noncoding RNAs in development and stem cells. Development 2016, 143, 3882–3894. [Google Scholar] [CrossRef] [Green Version]
- Hubler, M.J.; Kennedy, A.J. Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem. 2016, 34, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Hassan, H.; Hollingsworth, M.A.; Clausen, H. A novel human UDP-N-acetyl-D-galactosamine: Polypeptide N-acetylgalactosaminyltransferase, GalNAc-T7, with specificity for partial GalNAc-glycosylated acceptor substrates. FEBS Lett. 1999, 460, 226–230. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| READS | |
|---|---|
| Total reads | 856,863,196 |
| Mean reads per sample | 71,405,266 |
| Mean trimmed reads | 98.71% |
| ASSEMBLY | |
| Contigs | 219,765 |
| Minimum length | 200 bp |
| Maximum length | 16,164 bp |
| Average length | 493 bp |
| N50 | 539 bp |
| LncRNAs | |
| Potential lncRNAs | 19,168 (8.72%) |
| Average length | 499.54 bp |
| lncRNAs mapped on genome | 6234 (32.52%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereiro, P.; Moreira, R.; Novoa, B.; Figueras, A. Differential Expression of Long Non-Coding RNA (lncRNA) in Mediterranean Mussel (Mytilus galloprovincialis) Hemocytes under Immune Stimuli. Genes 2021, 12, 1393. https://doi.org/10.3390/genes12091393
Pereiro P, Moreira R, Novoa B, Figueras A. Differential Expression of Long Non-Coding RNA (lncRNA) in Mediterranean Mussel (Mytilus galloprovincialis) Hemocytes under Immune Stimuli. Genes. 2021; 12(9):1393. https://doi.org/10.3390/genes12091393
Chicago/Turabian StylePereiro, Patricia, Rebeca Moreira, Beatriz Novoa, and Antonio Figueras. 2021. "Differential Expression of Long Non-Coding RNA (lncRNA) in Mediterranean Mussel (Mytilus galloprovincialis) Hemocytes under Immune Stimuli" Genes 12, no. 9: 1393. https://doi.org/10.3390/genes12091393
APA StylePereiro, P., Moreira, R., Novoa, B., & Figueras, A. (2021). Differential Expression of Long Non-Coding RNA (lncRNA) in Mediterranean Mussel (Mytilus galloprovincialis) Hemocytes under Immune Stimuli. Genes, 12(9), 1393. https://doi.org/10.3390/genes12091393