
The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Data Collection
2.3. Genetic Analysis
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Clinical Examination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fiskerstrand, T.; H’Mida-Ben Brahim, D.; Johansson, S.; M’Zahem, A.; Haukanes, B.I.; Drouot, N.; Zimmermann, J.; Cole, A.J.; Vedeler, C.; Bredrup, C.; et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am. J. Hum. Genet. 2010, 87, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Fiskerstrand, T.; Knappskog, P.; Majewski, J.; Wanders, R.J.; Boman, H.; Bindoff, L.A. A novel Refsum-like disorder that maps to chromosome 20. Neurology 2009, 72, 20. [Google Scholar] [CrossRef] [PubMed]
- Savinainen, J.R.; Saario, S.M.; Laitinen, J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. (Oxf.) 2012, 204, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.; Ware, T.B.; Lee, H.-C.; Hsu, K.-L. Lipid-metabolizing serine hydrolases in the mammalian central nervous system: Endocannabinoids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Shaikh, M.; Singh, S.; Rajendran, A.; Mhetre, A.; Kamat, S.S. Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids. J. Biol. Chem. 2018, 293, 16953–16963. [Google Scholar] [CrossRef] [Green Version]
- Kamat, S.S.; Camara, K.; Parsons, W.H.; Chen, D.-H.; Dix, M.M.; Bird, T.D.; Howell, A.R.; Cravatt, B.F. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat. Chem. Biol. 2015, 11, 164–171. [Google Scholar] [CrossRef]
- Chen, D.-H.; Naydenov, A.; Blankman, J.L.; Mefford, H.C.; Davis, M.; Sul, Y.; Barloon, A.S.; Bonkowski, E.; Wolff, J.; Matsushita, M.; et al. Two novel mutations in ABHD12: Expansion of the mutation spectrum in PHARC and assessment of their functional effects. Hum. Mutat. 2013, 34, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Eisenberger, T.; Slim, R.; Mansour, A.; Nauck, M.; Nürnberg, G.; Nürnberg, P.; Decker, C.; Dafinger, C.; Ebermann, I.; Bergmann, C.; et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J. Rare Dis. 2012, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, K.M.; Avila-Fernandez, A.; van Huet, R.A.C.; Corton, M.; Pérez-Carro, R.; Martín-Garrido, E.; López-Molina, M.I.; Blanco-Kelly, F.; Hoefsloot, L.H.; van Zelst-Stams, W.A.; et al. Exome Sequencing Extends the Phenotypic Spectrum for ABHD12 Mutations: From Syndromic to Nonsyndromic Retinal Degeneration. Ophthalmology 2014, 121, 1620–1627. [Google Scholar] [CrossRef]
- Thimm, A.; Rahal, A.; Schoen, U.; Abicht, A.; Klebe, S.; Kleinschnitz, C.; Hagenacker, T.; Stettner, M. Genotype-phenotype correlation in a novel ABHD12 mutation underlying PHARC syndrome. J. Peripher. Nervous Syst. 2020, 25, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Igelman, A.D.; Ku, C.; da Palma, M.M.; Georgiou, M.; Schiff, E.R.; Lam, B.L.; Sankila, E.-M.; Ahn, J.; Pyers, L.; Vincent, A.; et al. Expanding the clinical phenotype in patients with disease causing variants associated with atypical Usher syndrome. Ophthalmic Genet. 2021, 1–10. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Fishman, G.A.; Alexander, K.R.; Anderson, R.J.; Derlacki, D.J. Visual Acuity Impairment in Patients with Retinitis Pigmentosa. Ophthalmology 1996, 103, 1593–1600. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Thomas, H.B.; Rowlands, C.; Arno, G.; Beaman, G.; Gomes-Silva, B.; Campbell, C.; Gossan, N.; Hardcastle, C.; Webb, K.; et al. Functional and in-silico interrogation of rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. bioRxiv 2019, 781088. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Althubaiti, A. Information bias in health research: Definition, pitfalls, and adjustment methods. J. Multidiscip. Healthc. 2016, 9, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Blankman, J.L.; Long, J.Z.; Trauger, S.A.; Siuzdak, G.; Cravatt, B.F. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad. Sci. USA 2013, 110, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, H.; Hashimoto, T.; Murata, T.; Fukushima, K.; Sugaya, A.; Nishio, S.-y.; Usami, S.-i. Novel ABHD12 Mutations in PHARC Patients: The Differential Diagnosis of Deaf-Blindness. Ann. Otol. Rhinol. Laryngol. 2015, 124, 77S–83S. [Google Scholar] [CrossRef] [PubMed]
- Frasquet, M.; Lupo, V.; Chumillas, M.J.; Vázquez-Costa, J.F.; Espinós, C.; Sevilla, T. Phenotypical features of two patients diagnosed with PHARC syndrome and carriers of a new homozygous mutation in the ABHD12 gene. J. Neurol. Sci. 2018, 387, 134–138. [Google Scholar] [CrossRef]
- O’Malley, J.T.; Nadol, J.B., Jr.; McKenna, M.J. Anti CD163+, Iba1+, and CD68+ Cells in the Adult Human Inner Ear: Normal Distribution of an Unappreciated Class of Macrophages/Microglia and Implications for Inflammatory Otopathology in Humans. Otol. Neurotol. 2016, 37, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Mahajan, V.B.; Tsang, S.H. Rates of Bone Spicule Pigment Appearance in Patients With Retinitis Pigmentosa Sine Pigmento. Am. J. Ophthalmol. 2018, 195, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Possin, D.E.; Milam, A.H. Histopathology of Bone Spicule Pigmentation in Retinitis Pigmentosa. Ophthalmology 1995, 102, 805–816. [Google Scholar] [CrossRef]
- Nguyen, X.-T.-A.; Talib, M.; van Schooneveld, M.J.; Brinks, J.; Ten Brink, J.; Florijn, R.J.; Wijnholds, J.; Verdijk, R.M.; Bergen, A.A.; Boon, C.J.F. RPGR-Associated Dystrophies: Clinical, Genetic, and Histopathological Features. Int. J. Mol. Sci. 2020, 21, 835. [Google Scholar] [CrossRef] [Green Version]
- Pfau, M.; Jolly, J.K.; Wu, Z.; Denniss, J.; Lad, E.M.; Guymer, R.H.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S. Fundus-controlled perimetry (microperimetry): Application as outcome measure in clinical trials. Prog. Retin. Eye Res. 2020, 82, 100907. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M.; International Society For Clinical Electrophysiology of Vision. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. Adv. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Garway-Heath, D.; Rosen, P.; Bird, A.C.; Tuft, S.J. Outcome of cataract surgery in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2001, 85, 936. [Google Scholar] [CrossRef] [Green Version]
- Fishman, G.A.; Anderson, R.J.; Lourenco, P. Prevalence of posterior subcapsular lens opacities in patients with retinitis pigmentosa. Br. J. Ophthalmol. 1985, 69, 263. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Murakami, Y.; Nakatake, S.; Fujiwara, K.; Notomi, S.; Hisatomi, T.; Ishibashi, T. Factors Affecting Visual Acuity after Cataract Surgery in Patients with Retinitis Pigmentosa. Ophthalmology 2015, 122, 903–908. [Google Scholar] [CrossRef]
- Hong, Y.; Li, H.; Sun, Y.; Ji, Y. A Review of Complicated Cataract in Retinitis Pigmentosa: Pathogenesis and Cataract Surgery. J. Ophthalmol. 2020, 2020, 6699103. [Google Scholar] [CrossRef]
- Bell, S.J.; Oluonye, N.; Harding, P.; Moosajee, M. Congenital cataract: A guide to genetic and clinical management. Ther. Adv. Rare Dis. 2020, 1, 2633004020938061. [Google Scholar] [CrossRef]
- Gupta, A.; Ruminski, D.; Jimenez Villar, A.; Duarte Toledo, R.; Manzanera, S.; Panezai, S.; Mompean, J.; Artal, P.; Grulkowski, I. In vivo SS-OCT imaging of crystalline lens sutures. Biomed. Opt. Express 2020, 11, 5388–5400. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol 2019, 10, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation–Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A.; et al. Long-Term Structural Outcomes of Late-Stage RPE65 Gene Therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Family-ID | Sex, Age | Genetic Analysis | Presence of PHARC Syndrome Symptoms and Age at Symptom Onset/Diagnosis (Years) | |||||
|---|---|---|---|---|---|---|---|---|
| Allele 1/Allele 2 | Protein Change | Polyneuropathy | Hearing Loss | Ataxia | Retinitis Pigmentosa | Cataract | ||
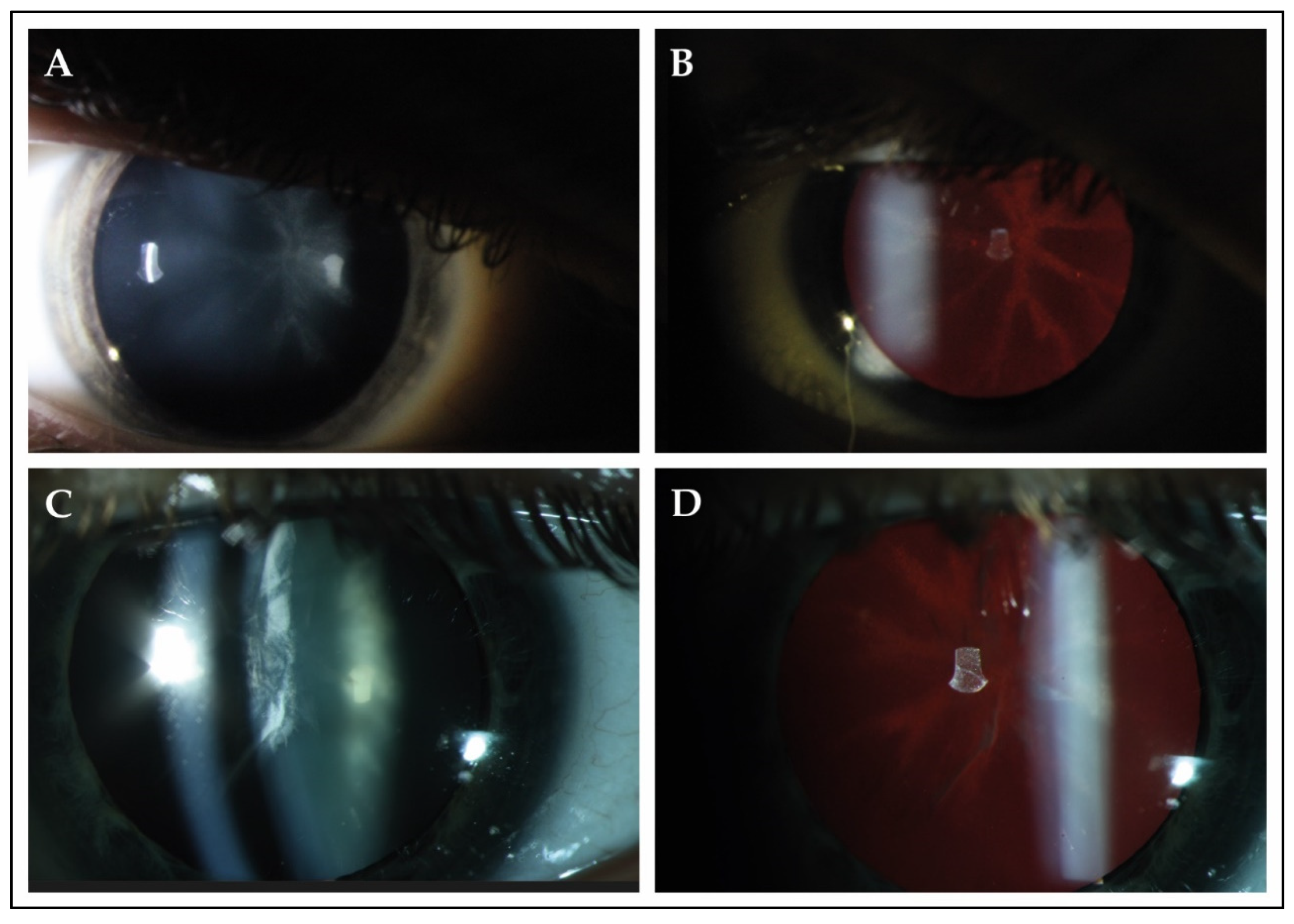
| A-1 | M, 47 | c.337_338delGAinsTTT/ c.1075del | p. (Asp113Phefs*15)/p. (Val359Phefs*27) | Pes cavus, hammertoes, distal sensory loss and absent tendon reflexes; age 8 | Yes; age 28 | Yes; age 8 | Asymptomatic, detected during electrophysiological testing at age 45 | Yes; age 36 |
| B-2 | F, 32 | c.337_338delGAinsTTT/ c.337_338delGAlinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | Yes; childhood | Yes; age 17 | Yes; age 45 | Reduced visual acuity; age 32 | Posterior subcapsular cataract; age 32 |
| C-3 * | M, 33 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Asymptomatic; but detected during examination at age 27 | No ¶ | Yes; age 27 | Night blindness; age 14 | Sutural cataract; age 3 |
| C-4 * | M, 33 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Distal muscle weakness and sensory loss; childhood | Yes; NA | Yes; age 27 | Night blindness; age 21 | Sutural cataract; age 3 |
| C-5 * | M, 38 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Abnormal gait pattern; childhood | Yes, 20 | Yes; age 31 | Night blindness | Star-shaped cataract; age 4 |
| D-6 | M, 42 | c.477G > A/c.557G > C | p. (Trp159*)/p. (Arg186Pro) | Distal sensory loss and reduced tendon reflexes; age 35 | Yes; age 36 | Yes; NA | Reduced visual acuity; age 29 | Cortical cataract; age 29 |
| E-7 † | F, 36 | c.337_338delGAinsTTT/ c.337_338delGAlinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | NA ‡ | Yes; age 12 | Yes; NA | Visual field loss; age 31 | Posterior subcapsular cataract; age 32 |
| F-8 | M, 53 | c.784C > T/c.867 + 5G > A | p. (Arg262*)/ p. (?) | Distal sensory loss; age 53 ‡ | Yes; age 20 | NA | Reduced visual acuity; age 18 | No |
| G-9 | M, 34 | c.620-2A > G/c.620-2A > G | p. (?)/p. (?) | Lower limb muscle weakness; age 31 ‡ | Yes; age 20 | NA | Reduced visual acuity and night blindness; age 22 | Yes; age 26 |
| H-10 † | M, 22 | c.193C > T/c.193 C > T | p. (Arg65*)/p. (Arg65*) | Lack of coordination; age 7 ‡ | No ¶ | NA | Reduced visual acuity; age 16 | No |
| I-11 † | M, 53 | c.374C > T/c.1154T > C | p. (Thr125Met)/p. (Leu385Pro) | NA, but epilepsy and learning difficulties ‡ | Yes; age 44 | NA | Reduced visual acuity and night blindness; age 30 | Posterior polar cataract; age 41 |
| J-12 * | M, 20 | c.337_338delGAinsTTT/c.337_338delGAinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | Yes; age 20 | Yes; age 16 | No | Night blindness; age 16 | Star-shaped cataract; age 17 |
| J-13 * | M, 17 | c.337_338delGAinsTTT/c.337_338delGAinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | Yes; age 18 | Yes; age 10 | No | Reduced visual acuity; age 10 | Star-shaped cataract; age 10 |
| K-14 | F, 46 | c.1063C > T/c.1063C > T | p. (Arg355*)/p. (Arg355*) | Yes; age 47 | Yes; NA | Yes; NA | Yes; NA | Cerulean cataract, NA |
| L-15 | M, 39 | c.337_338delGAinsTTT/c.341dup | p. (Asp113Phefs*15)/p. (Leu114Phefs*14) | NA ‡ | Yes; age 33 | NA | Night blindness; age 23 | Star-shaped cataract; age 29 |
| Family-ID | Sex, Age | BCVA (OD; OS) | Lens Status; Age at First Surgery | ffERG | Fundus Findings | |||
|---|---|---|---|---|---|---|---|---|
| Macular Changes | Bone Spicules | Spectral-Domain Optical Coherence Tomography | Fundus Autofluorescence | |||||
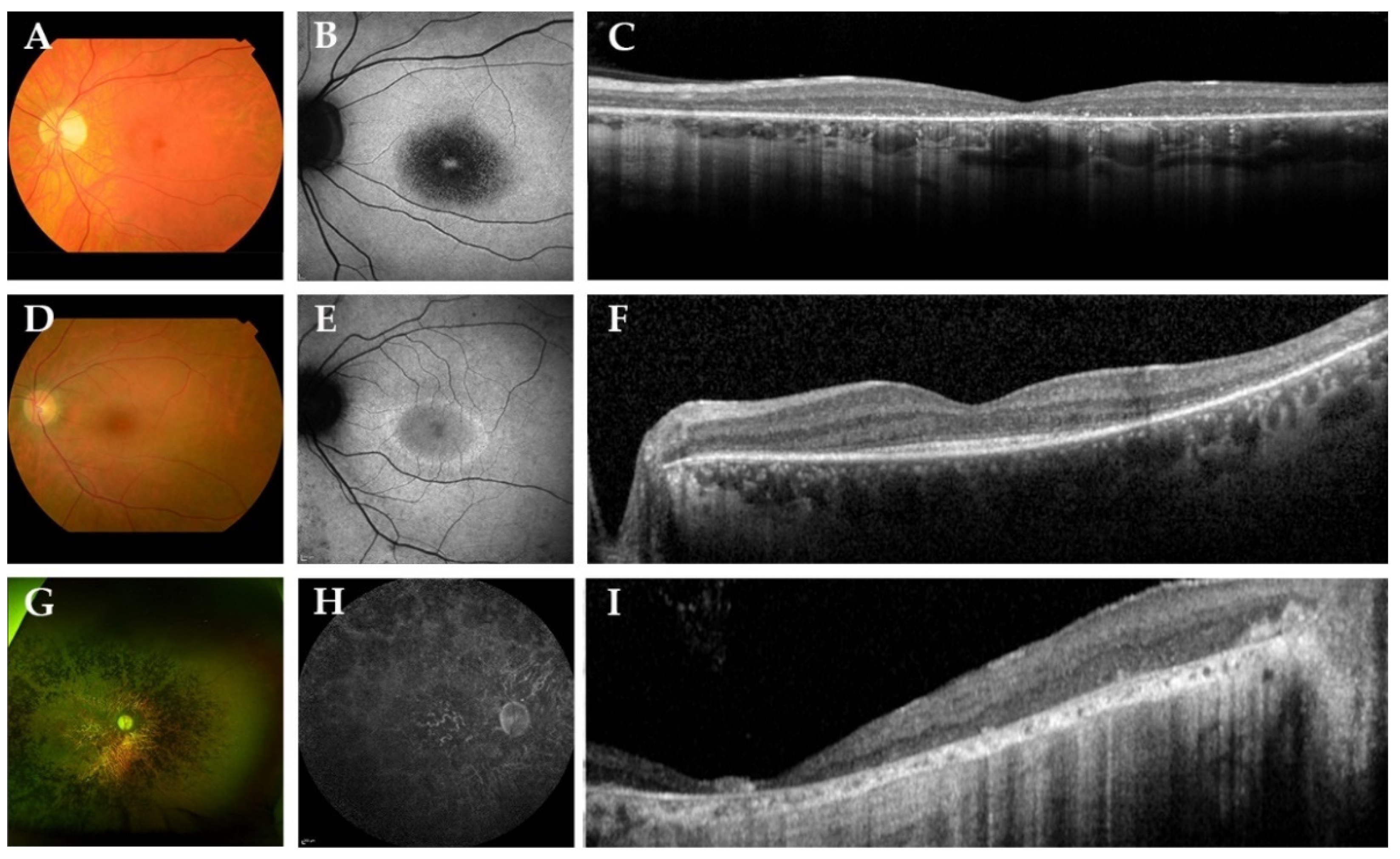
| A-1 | M, 47 | 20/22; 20/22 | Pseudophakic; surgery at age 36 | RCD | RPE alterations | No | Epiretinal membrane, degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Hypo-AF regions in midperiphery with a macular hyper-AF ring |
| B-2 | F, 32 | 20/25; 20/25 | Pseudophakic; surgery at age 32 | RCD | RPE alterations | No | Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Central hypo-AF surrounded by a hyper-AF ring |
| C-3 | M, 33 | 20/200; 20/200 | Pseudophakic; surgery at age 26 | NA | Atrophy | Yes | Degeneration of the outer retina | NA |
| C-4 | M, 33 | 20/125; 20/100 | Pseudophakic; surgery at age 21 | MR | Atrophy | Yes | Epiretinal membrane, degeneration of the outer retina, CME ODS at age 29, resolved at age 31 | Hypo-AF lesions in the midperiphery with hyper-AF changes in the central macula |
| C-5 | M, 38 | 20/134; 20/134 | Pseudophakic; surgery at age 19 | MR | Atrophy | Yes | Epiretinal membrane, degeneration of the outer retina, CME ODS at age 30, resolved at age 32 | NA |
| D-6 | M, 42 | 20/400; 20/400 | Cortical cataract | RCD | Atrophy | No | Degeneration of the outer retina | Central hypo-AF with a hyper-AF foveal spot |
| E-7 | F, 36 | 20/29; 20/29 | Pseudophakic; surgery at age 29 | RCD | RPE alterations | No | Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Hypo-AF regions in midperiphery with a macular hyper-AF ring |
| F-8 | M, 53 | LP; LP | Clear lens | NA | Atrophy | Yes | Extensive atrophy of all retinal layers | Generalized hypo-AF |
| G-9 | M, 34 | 20/400; 20/400 | Pseudophakic; surgery at age 34 | RCD | Atrophy | No | Extensive atrophy of all retinal layers at the fovea, with relatively preserved layers in the perifovea | Central hypo-AF |
| H-10 | M, 22 | 20/240; 20/240 | Clear lens | RCD | Bull’s eye | No | Degeneration of the outer retina | Central hypo-AF |
| I-11 | M, 53 | HM; HM | Pseudophakic; surgery at age 44 | NA | Atrophy and Macular hole OS | Yes | Degeneration of the outer retina. Macular hole OS. | Mottled patches of hypo-AF in nasal region with hypo-AF in the central macula |
| J-12 | M, 20 | 20/200; 20/200 | Pseudophakic; surgery at age 20 | NA | Atrophy | No | Degeneration of the outer retina. | Central hypo-AF with hyper-AF borders |
| J- 13 | M, 17 | 20/50; 20/40 | Star-shaped cataract | NA | Atrophy | No | Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Hyper-AF ring surrounded by a larger hyper-AF ring |
| K-14 | F, 46 | HM; 20/400 | Cerulean cataract | RCD | Atrophy | Yes | Degeneration of the outer retina. | Central hypo-AF with a hyper-AF foveal spot. Several hypo-AF lesions along the superior vascular arcade. |
| L-15 | M, 39 | 20/134; 20/200 | Pseudophakic; surgery at age 39 | RCD | Atrophy | Yes | Degeneration of the outer retina. CME ODS at age 33, resolved at age 39 | Generalized hypo-AF with preserved AF in the central macula. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, X.-T.-A.; Almushattat, H.; Strubbe, I.; Georgiou, M.; Li, C.H.Z.; van Schooneveld, M.J.; Joniau, I.; De Baere, E.; Florijn, R.J.; Bergen, A.A.; et al. The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective. Genes 2021, 12, 1404. https://doi.org/10.3390/genes12091404
Nguyen X-T-A, Almushattat H, Strubbe I, Georgiou M, Li CHZ, van Schooneveld MJ, Joniau I, De Baere E, Florijn RJ, Bergen AA, et al. The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective. Genes. 2021; 12(9):1404. https://doi.org/10.3390/genes12091404
Chicago/Turabian StyleNguyen, Xuan-Thanh-An, Hind Almushattat, Ine Strubbe, Michalis Georgiou, Catherina H. Z. Li, Mary J. van Schooneveld, Inge Joniau, Elfride De Baere, Ralph J. Florijn, Arthur A. Bergen, and et al. 2021. "The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective" Genes 12, no. 9: 1404. https://doi.org/10.3390/genes12091404
APA StyleNguyen, X. -T. -A., Almushattat, H., Strubbe, I., Georgiou, M., Li, C. H. Z., van Schooneveld, M. J., Joniau, I., De Baere, E., Florijn, R. J., Bergen, A. A., Hoyng, C. B., Michaelides, M., Leroy, B. P., & Boon, C. J. F. (2021). The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective. Genes, 12(9), 1404. https://doi.org/10.3390/genes12091404