
Environmental Influences Measured by Epigenetic Clock and Vulnerability Components at Birth Impact Clinical ASD Heterogeneity

 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Risk Factor Variables
2.3. Statistical Analysis
2.4. Methylation Analysis
2.5. Enrichment Analyses rVarbase and VMRs
2.6. Whole-Exome Analysis
2.7. Regression Models
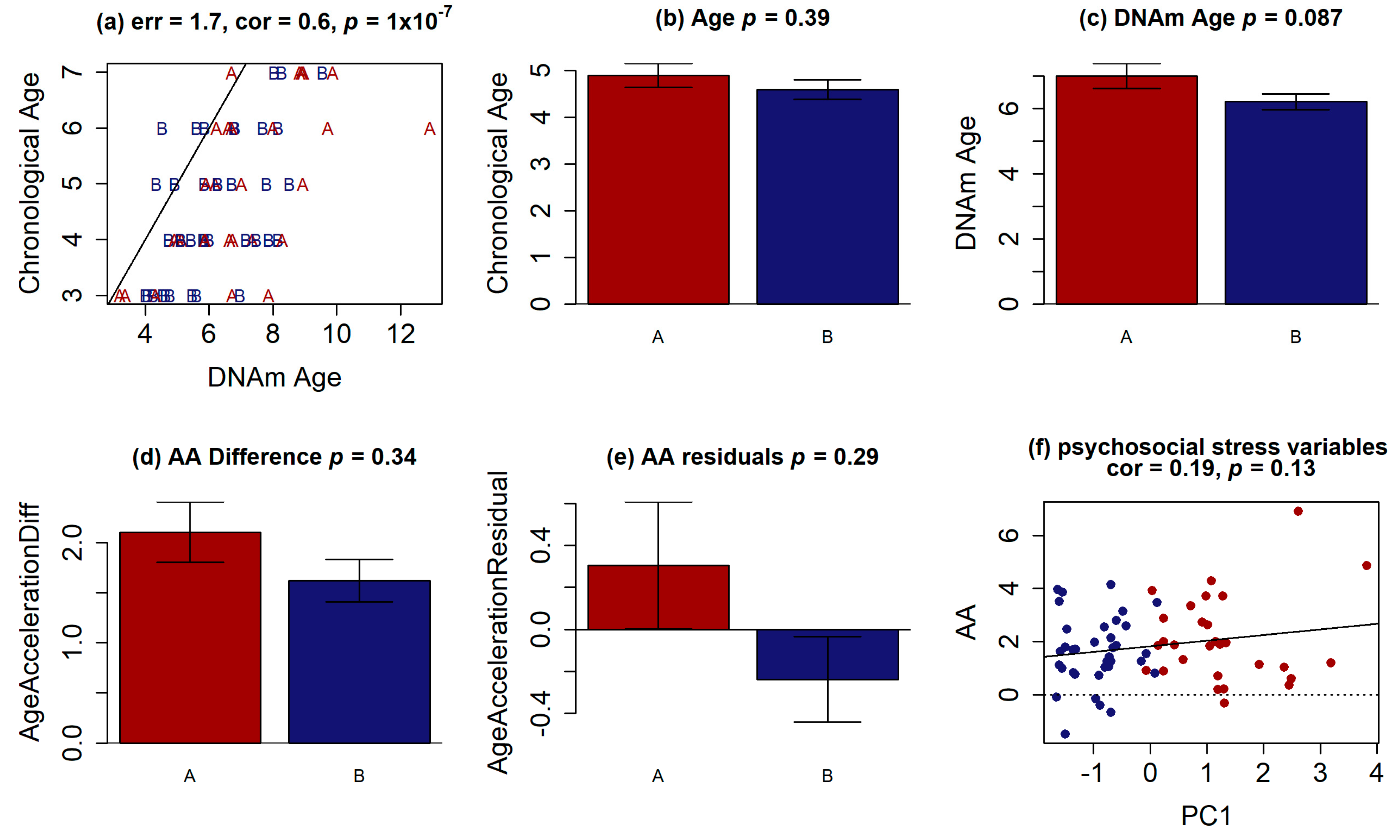
3. Results
3.1. Participants Characteristics
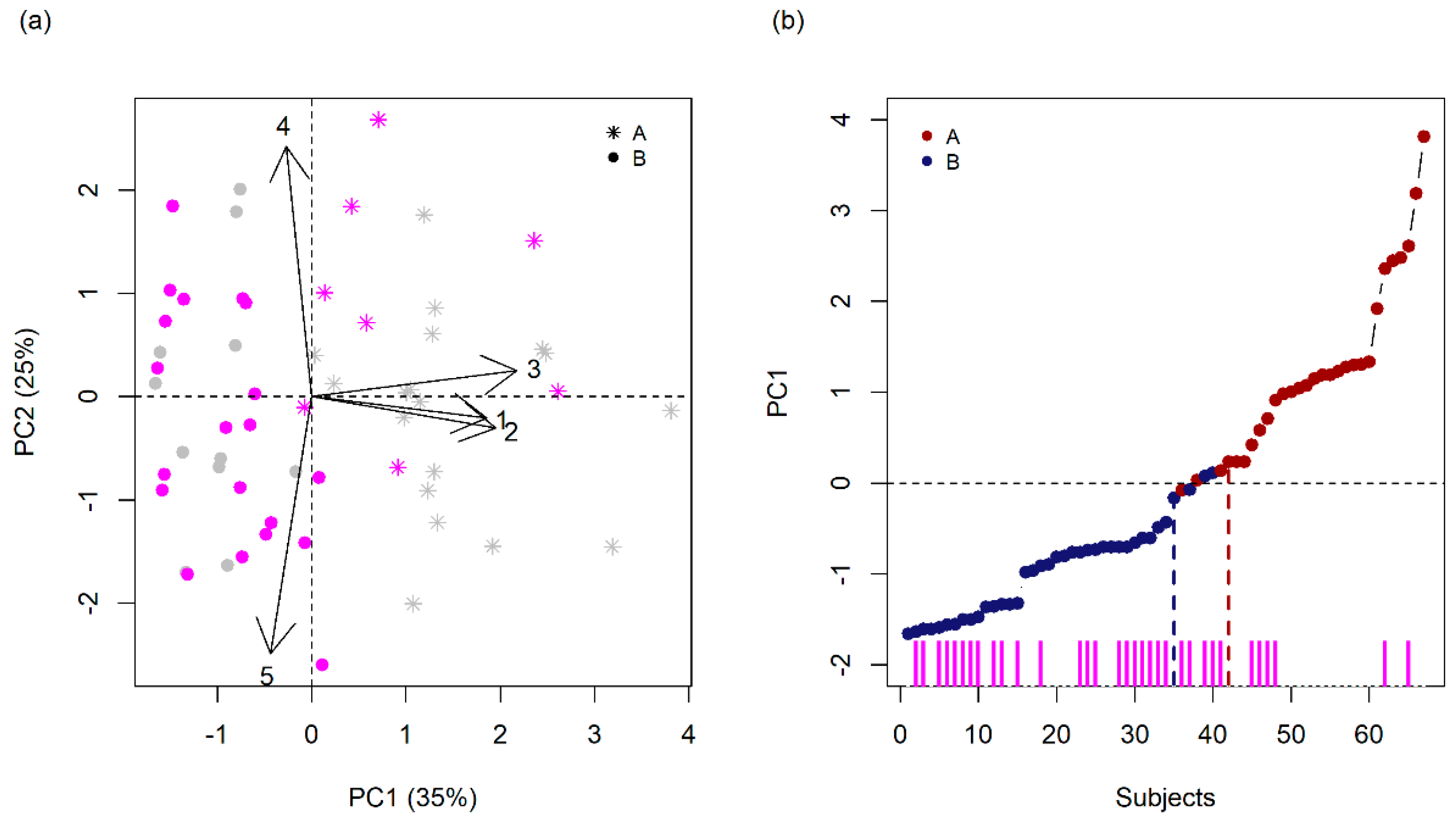
3.2. Finding Components of Vulnerability
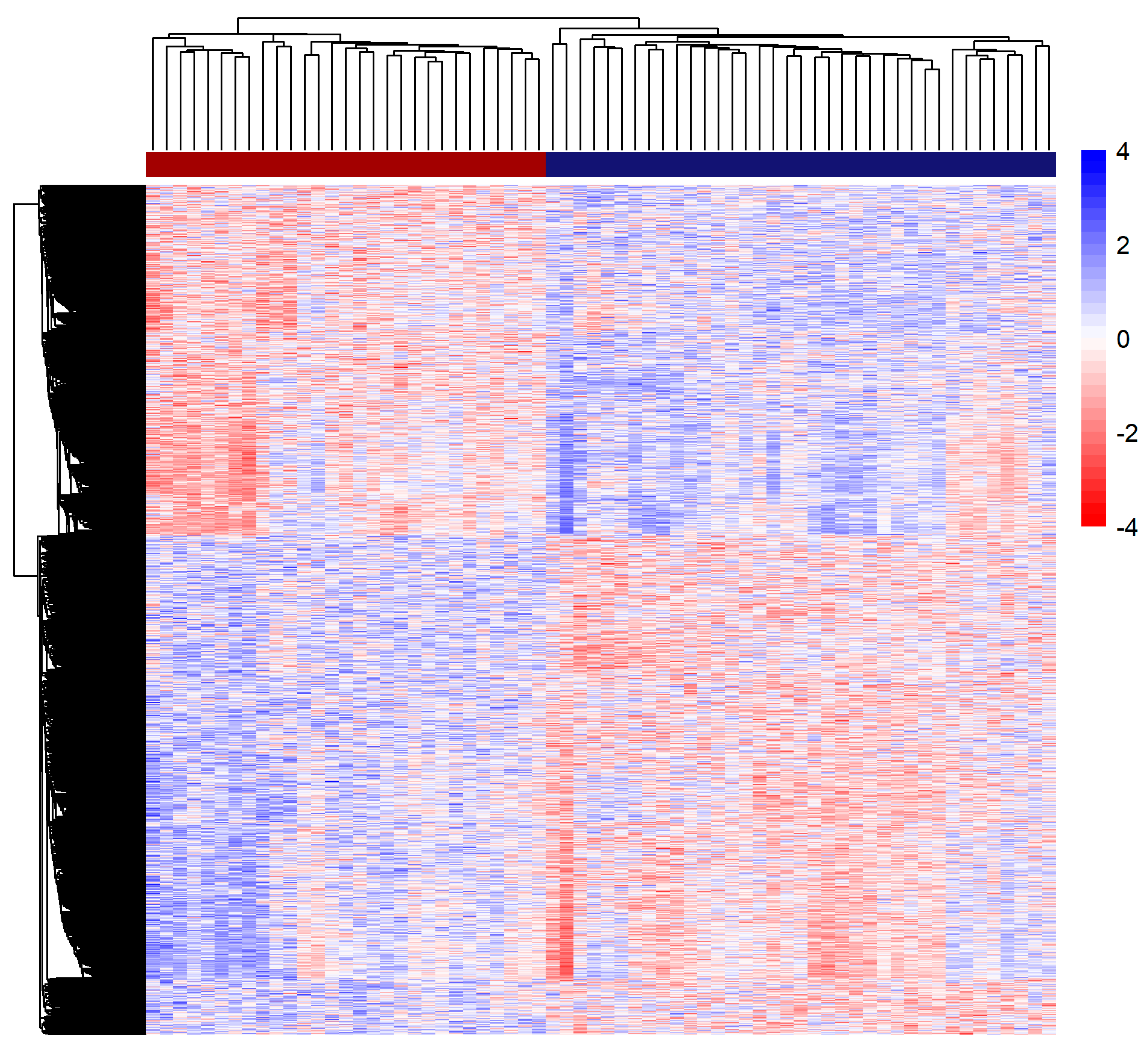
3.3. Methylation Analysis
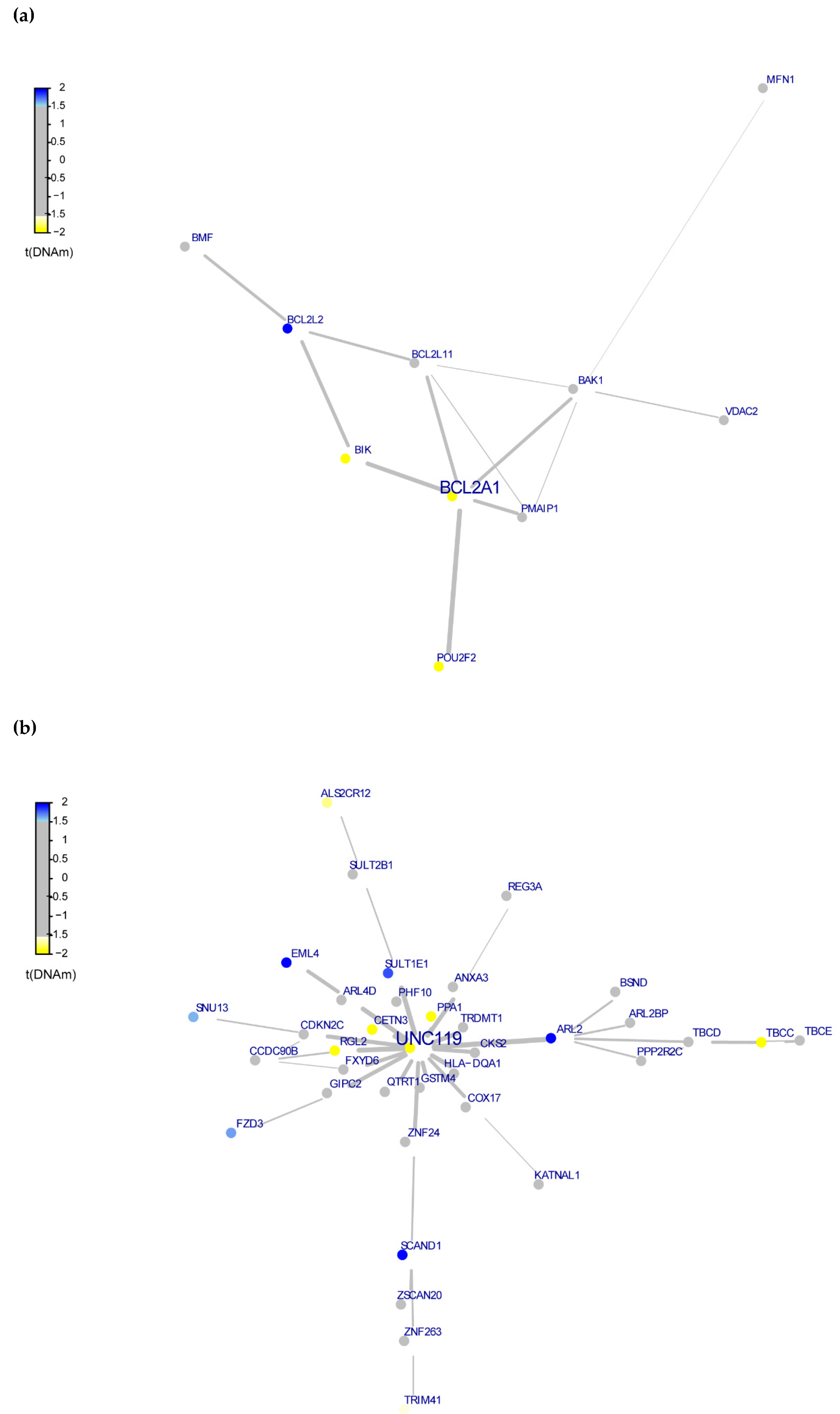
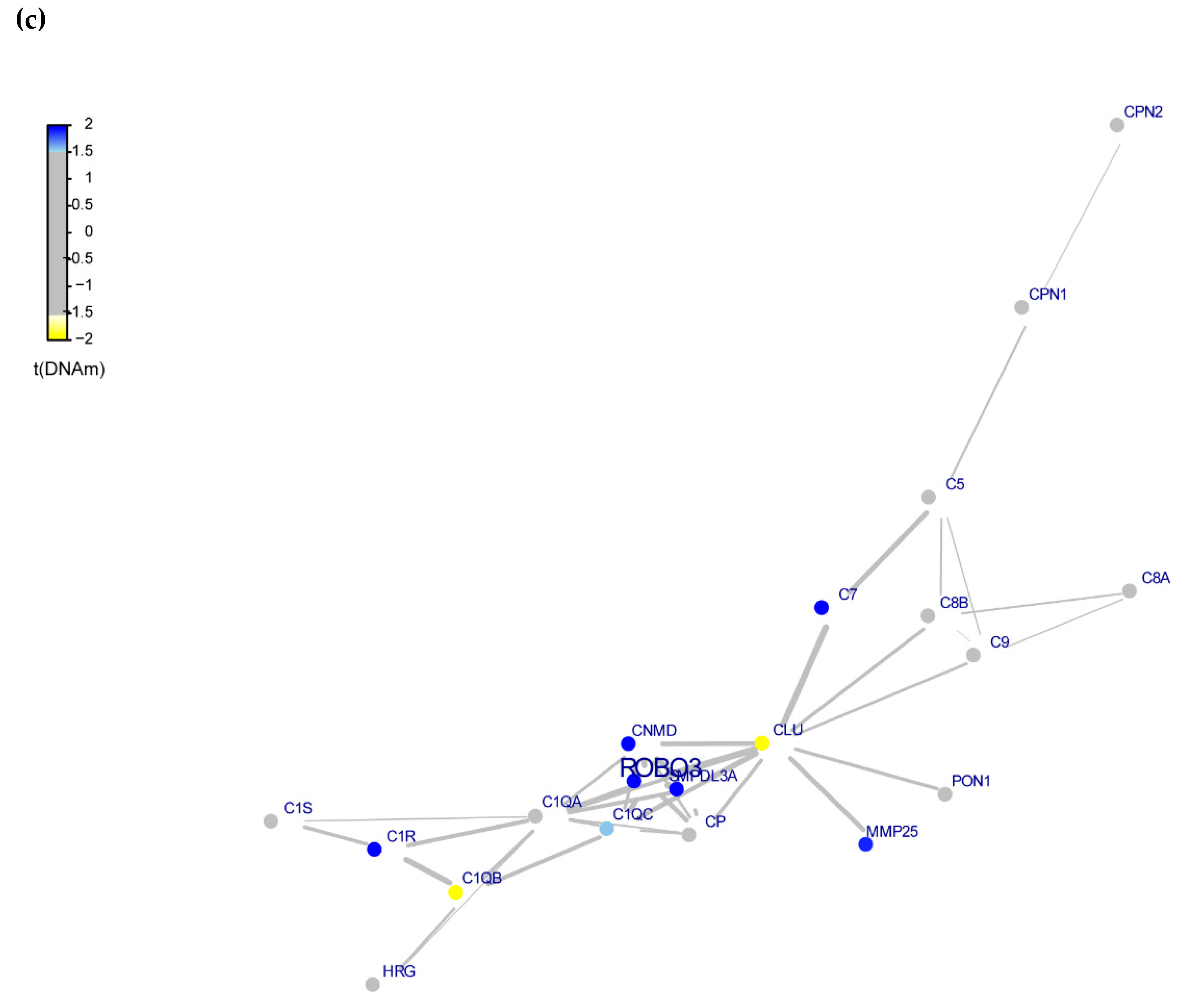
3.4. Whole-Exome Analysis
3.5. Regression Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B. VMRs Dataset Details
- Islam et al. [23] with a systematic comparison of genome-wide DNAm patterns (DNAm variability, cross-tissue DNAm concordance, and genetic determinants of DNAm across two independent early life cohorts encompassing different ages) between matched pediatric buccal epithelial cells (BECs) and peripheral blood mononuclear cells (PBMCs). They overlapped CpGs that were identified as (a) informative (i.e., variable across individuals and correlated between BECs and PBMCs) (8140), (b) differentially methylated between matched tissues (139,662), or (c) under genetic influence (4980; i.e., number of unique CpGs associated with validated cismQTL across two cohorts;
- Teh et al. [20], an examination of the relative influences of genotypic, environmental, and gene x environment interactive effects on the neonatal methylome. They studied the variation in genome-wide DNA methylation patterns in umbilical cord samples, genotyping, and measures of in utero environmental conditions and identified 1423 interindividual variably methylated regions (VMRs) across the 237 individuals. Methylation levels at 25% of the 1423 VMR-CpGs were best explained by genotype alone, while the rest were best explained by G × E models.
- Garg et al. [25] performed a screen to identify regions of common epigenetic variation using population data derived from five different human cell types. They searched for clusters of probes with high inter-individual variability (VMRs) and explored the potential underlying factors associated with the regulation of VMRs using different strategies (VMRs influenced by genetic, environmental, and GxE effects), and
- Hachiya et al. [19] catalog of EWAS VMRs.
References
- Lai, M.-C.; Lombardo, M.V.; Chakrabarti, B.; Baron-Cohen, S. Subgrouping the Autism “Spectrum”: Reflections on DSM-5. PLoS Biol. 2013, 11, e1001544. [Google Scholar] [CrossRef]
- Ousley, O.; Cermak, T. Autism Spectrum Disorder: Defining Dimensions and Subgroups. Curr. Dev. Disord. Rep. 2014, 1, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, D.J.; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; Bybjerg-Grauholm, J.; et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratten, J.; Wray, N.R.; Keller, M.C.; Visscher, P.M. Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat. Neurosci. 2014, 17, 782–790. [Google Scholar] [CrossRef]
- Jokiranta, E.; Brown, A.S.; Heinimaa, M.; Cheslack-Postava, K.; Suominen, A.; Sourander, A. Parental psychiatric disorders and autism spectrum disorders. Psychiatry Res. 2013, 207, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Daniels, J.L.; Forssen, U.; Hultman, C.M.; Cnattingius, S.; Savitz, D.A.; Feychting, M.; Sparen, P. Parental Psychiatric Disorders Associated With Autism Spectrum Disorders in the Offspring. Pediatrics 2008, 121, e1357–e1362. [Google Scholar] [CrossRef] [Green Version]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415. [Google Scholar] [CrossRef]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Mochizuki, K. Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders. Int. J. Environ. Res. Public Health 2016, 13, 504. [Google Scholar] [CrossRef] [Green Version]
- Sealey, L.A.; Hughes, B.W.; Sriskanda, A.N.; Guest, J.R.; Gibson, A.D.; Johnson-Williams, L.; Pace, D.G.; Bagasra, O. Environmental factors in the development of autism spectrum disorders. Environ. Int. 2016, 88, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A.S. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- Ciernia, A.V.; LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 2016, 17, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.W.; Anderson, P.J.; Battin, M.; Bowen, J.R.; Brown, N.; Callanan, C.; Campbell, C.; Chandler, S.; Cheong, J.; Darlow, B.; et al. Long term follow up of high risk children: Who, why and how? BMC Pediatr. 2014, 14, 279. [Google Scholar] [CrossRef] [Green Version]
- Keil, K.P.; Lein, P.J. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenetics 2016, 2, dvv012. [Google Scholar] [CrossRef] [Green Version]
- de la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef]
- Hachiya, T.; Furukawa, R.; Shiwa, Y.; Ohmomo, H.; Ono, K.; Katsuoka, F.; Nagasaki, M.; Yasuda, J.; Fuse, N.; Kinoshita, K.; et al. Genome-wide identification of inter-individually variable DNA methylation sites improves the efficacy of epigenetic association studies. NPJ Genom. Med. 2017, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.-L.; Dogra, S.; Wong, J.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.-M.; et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014, 24, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.V.; Ellis, S.E.; Bakulski, K.M.; Sheppard, B.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Feinberg, A.P.; Arking, D.E.; Ladd-Acosta, C.; et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 2017, 8, 1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, E.; Spiers, H.; Viana, J.; Pidsley, R.; Burrage, J.; Murphy, T.M.; Troakes, C.; Turecki, G.; O’Donovan, M.C.; Schalkwyk, L.C.; et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci. 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.A.; Goodman, S.J.; MacIsaac, J.L.; Obradović, J.; Barr, R.G.; Boyce, W.T.; Kobor, M.S. Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform EWAS design and interpretation. Epigenetics Chromatin 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C.; Leitão, E.; Wallner, S.; Schmitz, G.; Klein-Hitpass, L.; Sinha, A.; Jöckel, K.-H.; Heilmann-Heimbach, S.; Hoffmann, P.; Nöthen, M.M.; et al. Regions of common inter-individual DNA methylation differences in human monocytes: Genetic basis and potential function. Epigenetics Chromatin 2017, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Joshi, R.S.; Watson, C.; Sharp, A.J. A survey of inter-individual variation in DNA methylation identifies environmentally responsive co-regulated networks of epigenetic variation in the human genome. PLoS Genet. 2018, 14, e1007707. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Grzadzinski, R.; Huerta, M.; Lord, C. DSM-5 and autism spectrum disorders (ASDs): An opportunity for identifying ASD subtypes. Mol. Autism 2013, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Cholemkery, H.; Medda, J.; Lempp, T.; Freitag, C.M. Classifying Autism Spectrum Disorders by ADI-R: Subtypes or Severity Gradient? J. Autism Dev. Disord. 2016, 46, 2327–2339. [Google Scholar] [CrossRef]
- James, W.H. A potential explanation of some established major risk factors for autism. Dev. Med. Child Neurol. 2012, 54, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.L.; Seltzer, M.M. Changes in the autism behavioral phenotype during the transition to adulthood. J. Autism Dev. Disord. 2010, 40, 1431–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.H.; Takahashi, T.N.; Bagby, S.; Sahota, P.K.; Vaslow, D.F.; Wang, C.H.; Hillman, R.E.; Farmer, J.E. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am. J. Med. Genet. A 2005, 135, 171–180. [Google Scholar] [CrossRef]
- Munson, J.; Dawson, G.; Sterling, L.; Beauchaine, T.; Zhou, A.; Koehler, E.; Lord, C.; Rogers, S.; Sigman, M.; Estes, A.; et al. Evidence for Latent Classes of IQ in Young Children With Autism Spectrum Disorder. Am. J. Ment. Retard. 2008, 113, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Virkud, Y.V.; Todd, R.D.; Abbacchi, A.M.; Zhang, Y.; Constantino, J.N. Familial aggregation of quantitative autistic traits in multiplex versus simplex autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef] [Green Version]
- Simpkin, A.J.; Hemani, G.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; Mcardle, W.L.; Ring, S.M.; Sharp, G.C.; Tilling, K.; Horvath, S.; et al. Prenatal and early life influences on epigenetic age in children: A study of mother–offspring pairs from two cohort studies. Hum. Mol. Genet. 2016, 25, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Agerbo, E.; Sullivan, P.F.; Vilhjálmsson, B.J.; Pedersen, C.B.; Mors, O.; Børglum, A.D.; Hougaard, D.M.; Hollegaard, M.V.; Meier, S.; Mattheisen, M.; et al. Polygenic Risk Score, Parental Socioeconomic Status, Family History of Psychiatric Disorders, and the Risk for Schizophrenia. JAMA Psychiatry 2015, 72, 635. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.-X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordini, D.; Paula, C.S.; Cunha, G.R.; Caetano, S.C.; Bagaiolo, L.F.; Ribeiro, T.C.; Martone, M.C.C.; Portolese, J.; Moya, A.C.; Brunoni, D.; et al. A randomised clinical pilot trial to test the effectiveness of parent training with video modelling to improve functioning and symptoms in children with autism spectrum disorders and intellectual disability. J. Intellect. Disabil. Res. 2020, 64, 629–643. [Google Scholar] [CrossRef]
- Becker, M.M.; Wagner, M.B.; Bosa, C.A.; Schmidt, C.; Longo, D.; Papaleo, C.; Riesgo, R.S. Translation and validation of Autism Diagnostic Interview-Revised (ADI-R) for autism diagnosis in Brazil. Arq. Neuropsiquiatr. 2012, 70, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Le Couteur, A.; Rutter, M.; Lord, C.; Rios, P.; Robertson, S.; Holdgrafer, M.; McLennan, J. Autism diagnostic interview: A standardized investigator-based instrument. J. Autism Dev. Disord. 1989, 19, 363–387. [Google Scholar] [CrossRef]
- Karino, C.A.; Laros, J.A.; Jesus, G.R. de Evidences of convergent validity of SON-R 2½-7[a] with WPPSI-III and WISC-III. Psicol. Reflexão e Crítica 2011, 24, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, S.; Sparrow, D. Vineland Adaptive Behavior Scales—Vineland II: Survey Forms Manual; Pearson Assessments: Bloomington, MN, USA, 2005. [Google Scholar]
- Schopler, E.; Reichler, R.J.; Renner, B.R. The Childhood Autism Rating Scale (CARS); Western Psychological Services: Beaverton, OR, USA, 1988; pp. 1–10. [Google Scholar]
- Schopler, E.; Reichler, R.J.; DeVellis, R.F.; Daly, K. Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J. Autism Dev. Disord. 1980, 10, 91–103. [Google Scholar] [CrossRef]
- Achenbach, T.M.; Rescorla, L.A. Child Behavior Checklist for Ages 6–18, Teacher’s Report Form, Youth Assessment, Self-Report: An Integrated System of Multi-Informant. 2001. Available online: https://store.aseba.org/MANUAL-FOR-THE-ASEBA-SCHOOL-AGE-FORMS-PROFILES/productinfo/505/ (accessed on 17 September 2021).
- Achenbach, T.M.; Resorta, L.A. Manual for ASEBA School-Age Forms and Profiles; ASEBA: Burlington, VT, USA, 2001; pp. 1–238. [Google Scholar]
- Carvalho, D.M.; Junqueira, G.P.; Gracioli, S.M.A.; Bordin, M.B.M. Avaliação do comportamento infantil: Uma revisão da literatura [Child behaviour assessment: A literature review]. In Anais do 3° Congresso de Iniciação Científica, 10° Encontro de Pesquisadoras da Uni-FACEF; Uni-FACEF: Franca, Brasil, 2009; Volume 3, pp. 164–174. [Google Scholar]
- Miller, G.E.; Chen, E.; Fok, A.K.; Walker, H.; Lim, A.; Nicholls, E.F.; Cole, S.; Kobor, M.S. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 14716–14721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delobel-Ayoub, M.; Ehlinger, V.; Klapouszczak, D.; Maffre, T.; Raynaud, J.P.; Delpierre, C.; Arnaud, C. Socioeconomic disparities and prevalence of autism spectrum disorders and intellectual disability. PLoS ONE 2015, 10, e0141964. [Google Scholar] [CrossRef] [PubMed]
- Brazilian Market Research Association. Brazilian Criteria 2015 and Social Class Distribution Update for 2016. ABEP—Associação Brasileira de Empresas de Pesquisa/Brazilian Market Research Association. 2016. Available online: https://www.abep.org/criterio-brasil (accessed on 11 August 2020).
- Kinney, D.K.; Miller, A.M.; Crowley, D.J.; Huang, E.; Gerber, E. Autism Prevalence Following Prenatal Exposure to Hurricanes and Tropical Storms in Louisiana. J. Autism Dev. Disord. 2008, 38, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.T.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef] [Green Version]
- Triche, T.J.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseman, E.A.; Molitor, J.; Marsit, C.J. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 2014, 30, 1431–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of β-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Widschwendter, M.; Teschendorff, A.E. A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 2014, 30, 2360–2366. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Du, Y.; Qu, S.; Wang, J. rVarBase: An updated database for regulatory features of human variants. Nucleic Acids Res. 2016, 44, D888–D893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger, B.E.; Saul, M.C.; Driessen, T.M.; Gammie, S.C. Development of a versatile enrichment analysis tool reveals associations between the maternal brain and mental health disorders, including autism. BMC Neurosci. 2013, 14, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Kamakura, W.; Mazzon, J.A. Socioeconomic stratification criteria and classification tools in Brazil. Rev. Adm. Empres. 2016, 56, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.D.; Jones, M.J.; Meaney, M.J.; Turecki, G.; Kobor, M.S. BECon: A tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 2017, 7, e1187. [Google Scholar] [CrossRef] [Green Version]
- Fernell, E.; Eriksson, M.; Gillberg, C. Early diagnosis of autism and impact on prognosis: A narrative review. Clin. Epidemiol. 2013, 5, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, B.S. Polygenic Scores in Epidemiology: Risk Prediction, Etiology, and Clinical Utility. Curr. Epidemiol. Rep. 2015, 2, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, A.M.; Hartman, C.A.; Franke, B.; Buitelaar, J.K.; Rommelse, N.N.J. Does the cognitive architecture of simplex and multiplex ASD families differ? J. Autism Dev. Disord. 2016, 46, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, E.; Zeller, C.; Masrour, N.; Patten, D.K.; Gallon, J.; Wilhelm-Benartzi, C.S.; Ghaem-Maghami, S.; Bowtell, D.D.; Brown, R. Genes Predisposed to DNA Hypermethylation during Acquired Resistance to Chemotherapy Are Identified in Ovarian Tumors by Bivalent Chromatin Domains at Initial Diagnosis. Cancer Res. 2018, 78, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Krook, M.A.; Hawkins, A.G.; Patel, R.M.; Lucas, D.R.; Van Noord, R.; Chugh, R.; Lawlor, E.R. A bivalent promoter contributes to stress-induced plasticity of CXCR4 in Ewing sarcoma. Oncotarget 2016, 7, 61775–61788. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.K.; Krakowiak, P.; Baker, A.; Hansen, R.L.; Ozonoff, S.; Hertz-Picciotto, I. Preeclampsia, Placental Insufficiency, and Autism Spectrum Disorder or Developmental Delay. JAMA Pediatr. 2015, 169, 154. [Google Scholar] [CrossRef]
- Connolly, N.; Anixt, J.; Manning, P.; Ping-I Lin, D.; Marsolo, K.A.; Bowers, K. Maternal metabolic risk factors for autism spectrum disorder-An analysis of electronic medical records and linked birth data. Autism Res. 2016, 9, 829–837. [Google Scholar] [CrossRef]
- Xie, S.; Karlsson, H.; Dalman, C.; Widman, L.; Rai, D.; Gardner, R.M.; Magnusson, C.; Schendel, D.E.; Newschaffer, C.J.; Lee, B.K. Family History of Mental and Neurological Disorders and Risk of Autism. JAMA Netw. Open 2019, 2, e190154. [Google Scholar] [CrossRef] [PubMed]
- Palma-Gudiel, H.; Córdova-Palomera, A.; Eixarch, E.; Deuschle, M.; Fañanás, L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: A meta-analysis. Epigenetics 2015, 10, 893–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Genetic Syndromes, Maternal Diseases and Antenatal Factors Associated with Autism Spectrum Disorders (ASD). Front. Neurosci. 2016, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Kalkbrenner, A.E.; Schmidt, R.J.; Penlesky, A.C. Environmental chemical exposures and autism spectrum disorders: A review of the epidemiological evidence. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 277–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilder, D.A.; Bakian, A.V.; Viskochil, J.; Clark, E.A.S.; Botts, E.L.; Smith, K.R.; Pimentel, R.; Mcmahon, W.M.; Coon, H. Maternal prenatal weight gain and autism spectrum disorders. Pediatrics 2013, 135, e1276–e1283. [Google Scholar] [CrossRef] [Green Version]
- Krakowiak, P.; Walker, C.K.; Tancredi, D.; Hertz-Picciotto, I.; Van de Water, J. Autism-specific maternal anti-fetal brain autoantibodies are associated with metabolic conditions. Autism Res. 2017, 10, 89–98. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | PC 1 | PC 2 | PC 3 | PC 4 | PC 5 |
|---|---|---|---|---|---|
| Variance (%) | 35.1 | 25.3 | 18.2 | 12.5 | 8.9 |
| Cumulative Variance (%) | 35.1 | 60.4 | 78.6 | 91.1 | 100 |
| Eigenvalue | 1.76 | 1.26 | 0.91 | 0.62 | 0.44 |
| Gestational Complications | −0.1 | 0.78 | −0.52 | 0.25 | −0.23 |
| Maternal Stress | 0.7 | −0.07 | −0.59 | −0.16 | 0.36 |
| Maternal Schooling | 0.74 | −0.1 | 0.24 | 0.62 | 0.02 |
| Social Class | 0.82 | 0.08 | 0.12 | −0.36 | −0.41 |
| Psychiatric Family History | −0.17 | −0.8 | −0.47 | 0.15 | −0.3 |
| Database | Total | Overlap | p-Value | FE | |
|---|---|---|---|---|---|
| Islam et al., 2019 [23] | Different tissues | 139,662 | 4549 | <1 × 10−4 | 1.23 |
| Informative | 8140 | 295 | <1 × 10−4 | 1.59 | |
| meQTL | 4980 | 143 | 0.16 | 1.09 | |
| Hachiya et al., 2017 [19] | VMRs EWAS | 269 | 17 | <1 × 10−4 | 2.31 |
| Garg et al., 2018 [25] | B-Cells | 4367 | 214 | <1 × 10−4 | 1.85 |
| Environmental | 804 | 27 | 0.12 | 1.28 | |
| Fibroblasts | 4788 | 149 | 0.05 | 1.15 | |
| Glia Cells | 6990 | 221 | 2.1 × 10−3 | 1.21 | |
| Neurons | 7075 | 230 | 1 × 10−4 | 1.27 | |
| T-Cells | 8940 | 396 | <1 × 10−4 | 1.72 | |
| Coeff. Estimation | Sd. Error | t-Value | Pr (>|t|) | |
|---|---|---|---|---|
| Intercept | 35.2 | 2.1 | 16.92 | <2 × 10−16 *** |
| AA | 0.33 | 0.55 | 0.6 | 0.55 |
| Group (0 = B, 1 = A) | −3.8 | 1.6 | −2.38 | 0.02 * |
| Sex (0 = female, 1 = male) | 3 | 2 | 1.48 | 0.14 |
| Model: Vineland~AA * PC1 * PC2 + Sex | ||||
|---|---|---|---|---|
| Coeff. Estimation | Sd. Error | t-Value | Pr (>|t|) | |
| Intercept | 44.4 | 2.6 | 16.75 | <2 × 10−16 *** |
| AA | 1.25 | 0.84 | 1.49 | 0.14 |
| PC1 | 2.2 | 1.2 | 1.82 | 0.07 |
| PC2 | 0.7 | 1.7 | 0.43 | 0.67 |
| Sex (0 = female, 1 = male) | 4 | 2.7 | 1.50 | 0.14 |
| AA:PC1 | −1.04 | 0.43 | −2.43 | 1.87 × 10−2 * |
| AA:PC2 | −1.63 | 0.79 | −2.05 | 4.49 × 10−2 * |
| PC1:PC2 | −0.7 | 1.4 | −0.52 | 0.6 |
| AA:PC1:PC2 | 0.85 | 0.84 | 1.00 | 0.32 |
| Model: Vineland~AA * PC1 * PC2 * PC3 + Sex | ||||
| Intercept | 42.15 | 2.51 | 16.80 | <2 × 10−16 *** |
| AA | 0.69 | 0.84 | 0.82 | 0.42 |
| PC1 | 1.11 | 1.11 | 1.00 | 0.32 |
| PC2 | 2.32 | 1.73 | 1.34 | 0.19 |
| PC3 | −9.12 | 2.89 | −3.16 | 2.82 × 10−3 ** |
| Sex (0 = female, 1 = male) | 7.71 | 2.70 | 2.85 | 6.46 × 10−3 ** |
| AA:PC1 | −0.47 | 0.43 | −1.10 | 0.28 |
| AA:PC2 | −2.62 | 0.84 | −3.11 | 3.19 × 10−3 ** |
| PC1:PC2 | −0.96 | 1.42 | −0.68 | 0.50 |
| AA:PC3 | 5.13 | 1.43 | 3.58 | 8.26 × 10−4 *** |
| PC1:PC3 | 3.39 | 1.95 | 1.74 | 0.09 |
| PC2:PC3 | −2.80 | 2.34 | −1.20 | 0.24 |
| AA:PC1:PC2 | 1.05 | 0.81 | 1.29 | 0.20 |
| AA:PC1:PC3 | −2.93 | 1.00 | −2.93 | 5.25 × 10−3 ** |
| AA:PC2:PC3 | 1.41 | 0.95 | 1.49 | 0.14 |
| PC1:PC2:PC3 | −0.97 | 2.16 | −0.45 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neri de Souza Reis, V.; Tahira, A.C.; Daguano Gastaldi, V.; Mari, P.; Portolese, J.; Feio dos Santos, A.C.; Lisboa, B.; Mari, J.; Caetano, S.C.; Brunoni, D.; et al. Environmental Influences Measured by Epigenetic Clock and Vulnerability Components at Birth Impact Clinical ASD Heterogeneity. Genes 2021, 12, 1433. https://doi.org/10.3390/genes12091433
Neri de Souza Reis V, Tahira AC, Daguano Gastaldi V, Mari P, Portolese J, Feio dos Santos AC, Lisboa B, Mari J, Caetano SC, Brunoni D, et al. Environmental Influences Measured by Epigenetic Clock and Vulnerability Components at Birth Impact Clinical ASD Heterogeneity. Genes. 2021; 12(9):1433. https://doi.org/10.3390/genes12091433
Chicago/Turabian StyleNeri de Souza Reis, Viviane, Ana Carolina Tahira, Vinícius Daguano Gastaldi, Paula Mari, Joana Portolese, Ana Cecilia Feio dos Santos, Bianca Lisboa, Jair Mari, Sheila C. Caetano, Décio Brunoni, and et al. 2021. "Environmental Influences Measured by Epigenetic Clock and Vulnerability Components at Birth Impact Clinical ASD Heterogeneity" Genes 12, no. 9: 1433. https://doi.org/10.3390/genes12091433
APA StyleNeri de Souza Reis, V., Tahira, A. C., Daguano Gastaldi, V., Mari, P., Portolese, J., Feio dos Santos, A. C., Lisboa, B., Mari, J., Caetano, S. C., Brunoni, D., Bordini, D., Silvestre de Paula, C., Vêncio, R. Z. N., Quackenbush, J., & Brentani, H. (2021). Environmental Influences Measured by Epigenetic Clock and Vulnerability Components at Birth Impact Clinical ASD Heterogeneity. Genes, 12(9), 1433. https://doi.org/10.3390/genes12091433