
p53 mRNA Metabolism Links with the DNA Damage Response

 , ,
, , 
Abstract
:1. Introduction
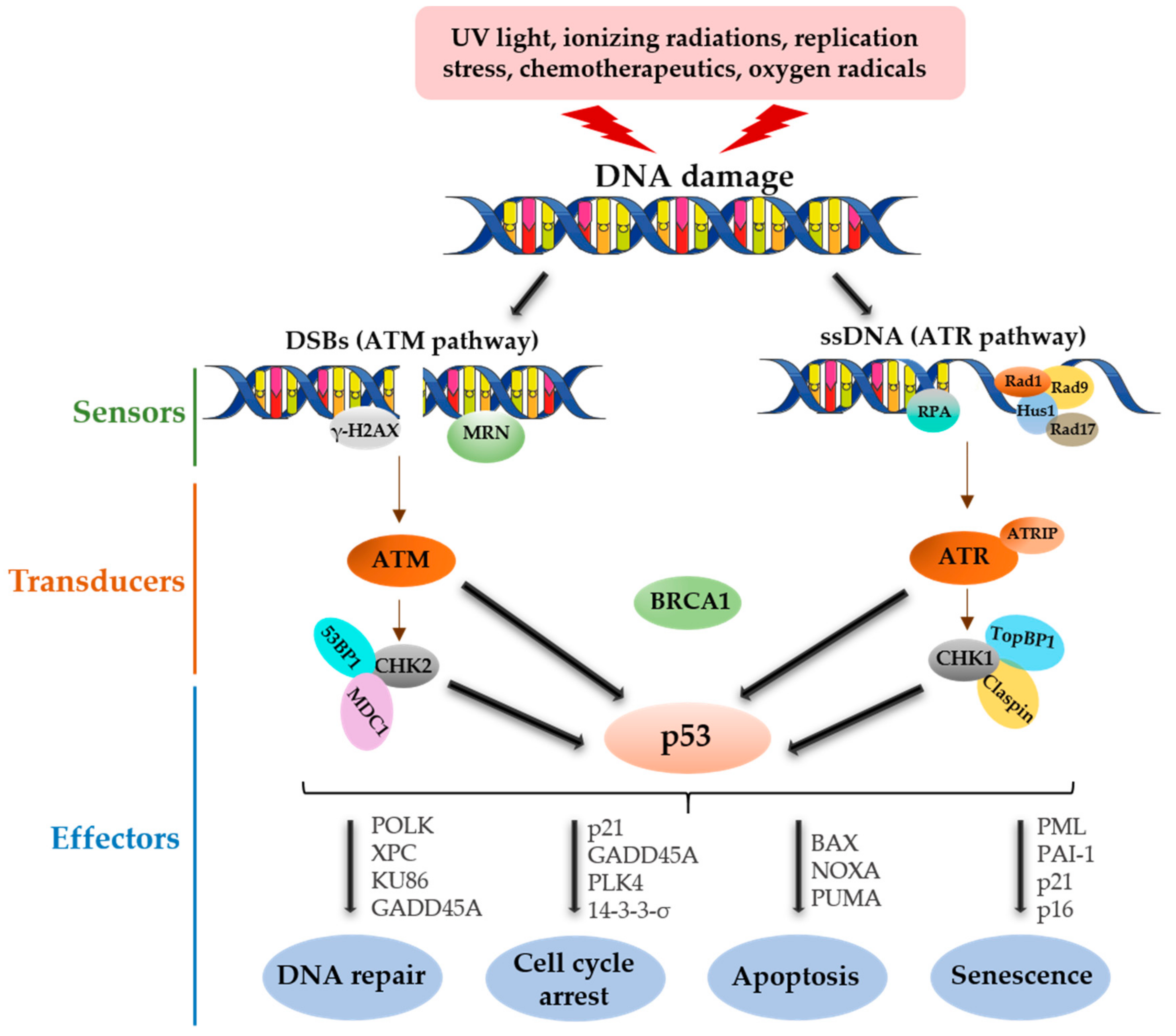
2. p53 Functional Roles in DNA Damage
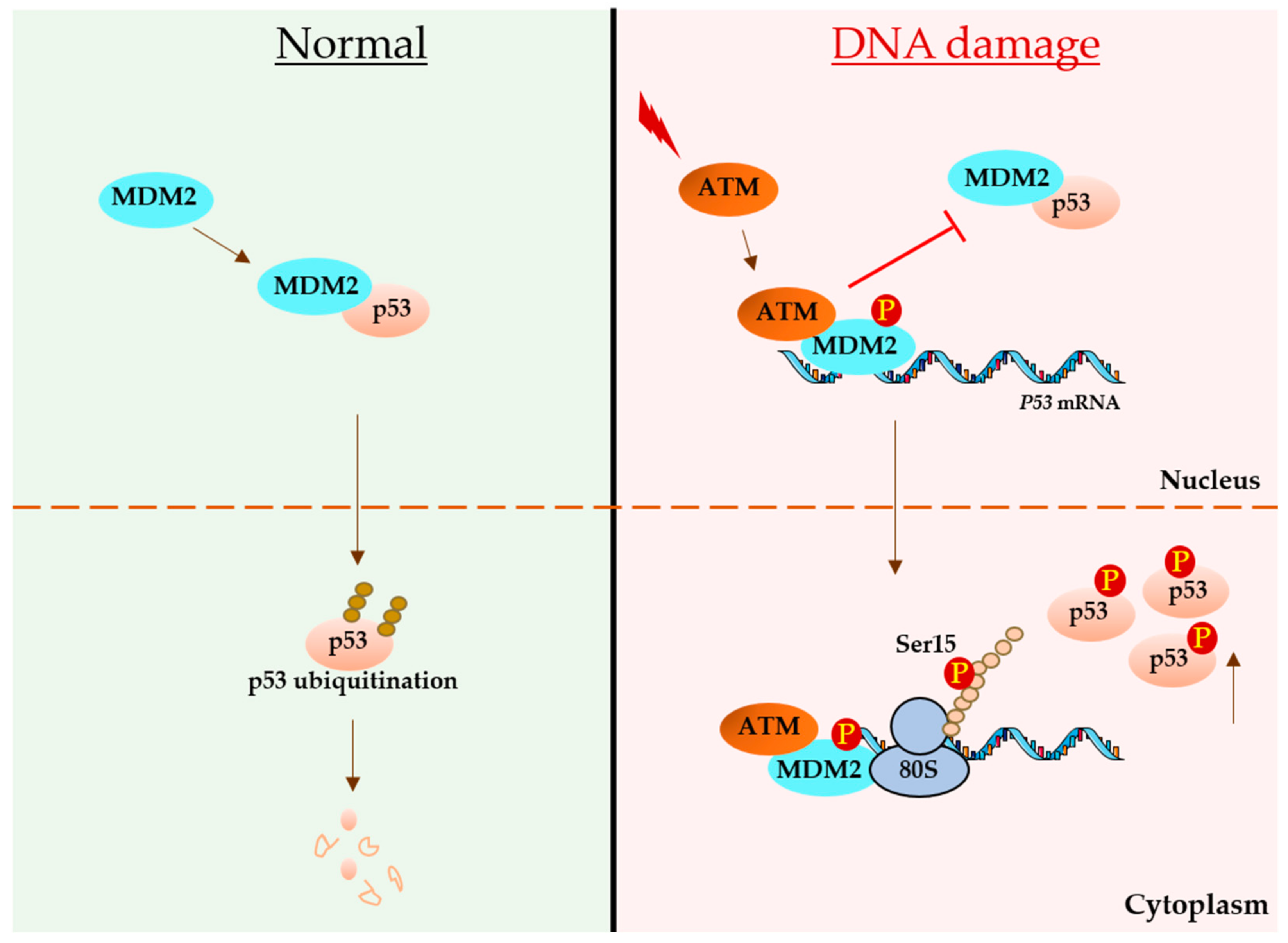
3. p53 mRNA Links with DNA Damage
3.1. Regulatory Roles of p53 Untranslated Regions (UTRs) during DDR
3.1.1. Roles of 5′ UTR
3.1.2. Roles of p53 3′ UTR
3.2. Regulatory Role of the p53-Coding Sequence during DNA Damage
3.3. Synonymous Mutations Regulating p53 Activation during DDR
4. Overview of RBPs Linked with the DDR and Functional Interplay with p53
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Halicka, H.D.; Darzynkiewicz, Z. Detection of histone H2AX phosphorylation on Ser-139 as an indicator of DNA damage (DNA double-strand breaks). Curr. Protoc. Cytom. 2004, 30, 7–27. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Burger, K.; Ketley, R.F.; Gullerova, M. Beyond the Trinity of ATM, ATR, and DNA-PK: Multiple Kinases Shape the DNA Damage Response in Concert With RNA Metabolism. Front. Mol. Biosci. 2019, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Speidel, D. The role of DNA damage responses in p53 biology. Arch. Toxicol. 2015, 89, 501–517. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Lazaro, M.; Fernandez-Gomez, F.J.; Jordan, J. p53: Twenty five years understanding the mechanism of genome protection. J. Physiol. Biochem. 2004, 60, 287–307. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fahraeus, R. The p53 mRNA: An integral part of the cellular stress response. Nucleic Acids Res. 2019, 47, 3257–3271. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Illana, V.; Fahraeus, R. p53 isoforms gain functions. Oncogene 2010, 29, 5113–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012, 22, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [Green Version]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, M.; Haessler, C.; Brandner, G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 1993, 8, 307–318. [Google Scholar] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [Green Version]
- Olivier, C.; Poirier, G.; Gendron, P.; Boisgontier, A.; Major, F.; Chartrand, P. Identification of a conserved RNA motif essential for She2p recognition and mRNA localization to the yeast bud. Mol. Cell Biol. 2005, 25, 4752–4766. [Google Scholar] [CrossRef] [Green Version]
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105. [Google Scholar] [CrossRef]
- Song, J.; Perreault, J.P.; Topisirovic, I.; Richard, S. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [Green Version]
- Lopez, I.; Tournillon, A.S.; Prado Martins, R.; Karakostis, K.; Malbert-Colas, L.; Nylander, K.; Fahraeus, R. p53-mediated suppression of BiP triggers BIK-induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ. 2017, 24, 1717–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournillon, A.S.; Lopez, I.; Malbert-Colas, L.; Findakly, S.; Naski, N.; Olivares-Illana, V.; Karakostis, K.; Vojtesek, B.; Nylander, K.; Fahraeus, R. p53 binds the mdmx mRNA and controls its translation. Oncogene 2017, 36, 723–730. [Google Scholar] [CrossRef]
- Gnanasundram, S.V.; Pyndiah, S.; Daskalogianni, C.; Armfield, K.; Nylander, K.; Wilson, J.B.; Fahraeus, R. PI3Kdelta activates E2F1 synthesis in response to mRNA translation stress. Nat. Commun. 2017, 8, 2103. [Google Scholar] [CrossRef] [Green Version]
- Vadivel Gnanasundram, S.; Fahraeus, R. Translation Stress Regulates Ribosome Synthesis and Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatkowska, A.; Dutkiewicz, M.; Zydowicz-Machtel, P.; Szpotkowska, J.; Janecki, D.M.; Ciesiolka, J. Translational Control in p53 Expression: The Role of 5′-Terminal Region of p53 mRNA. Int. J. Mol. Sci. 2019, 20, 5382. [Google Scholar] [CrossRef] [Green Version]
- Mosner, J.; Mummenbrauer, T.; Bauer, C.; Sczakiel, G.; Grosse, F.; Deppert, W. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 1995, 14, 4442–4449. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guo, K.; Kastan, M.B. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J. Biol. Chem. 2012, 287, 16467–16476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Kastan, M.B. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev. 2010, 24, 2146–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fahraeus, R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Caron de Fromentel, C.; Hainaut, P. p53 protein variants: Structural and functional similarities with p63 and p73 isoforms. Oncogene 2004, 23, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrable, H.; Sasaki, T.; Maier, B. DeltaNp53 or p44: Priming the p53 pump. Int. J. Biochem. Cell Biol. 2005, 37, 913–919. [Google Scholar] [CrossRef]
- Halaby, M.J.; Yang, D.Q. p53 translational control: A new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007, 395, 1–7. [Google Scholar] [CrossRef]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fahraeus, R. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.Q.; Halaby, M.J.; Zhang, Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 2006, 25, 4613–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedeken, L.; Singh, P.; Klempnauer, K.H. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J. Biol. Chem. 2011, 286, 42855–42862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cho, S.J.; Shu, L.; Yan, W.; Guerrero, T.; Kent, M.; Skorupski, K.; Chen, H.; Chen, X. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev. 2011, 25, 1528–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten-Gabbay, S.; Khan, D.; Liberman, N.; Yoffe, Y.; Bialik, S.; Das, S.; Oren, M.; Kimchi, A. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 2014, 33, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Li, Y.; Harris, B.R.; Jiang, S.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Translational Control Protein 80 Stimulates IRES-Mediated Translation of p53 mRNA in Response to DNA Damage. Biomed. Res. Int. 2015, 2015, 708158. [Google Scholar] [CrossRef] [Green Version]
- Lamaa, A.; Le Bras, M.; Skuli, N.; Britton, S.; Frit, P.; Calsou, P.; Prats, H.; Cammas, A.; Millevoi, S. A novel cytoprotective function for the DNA repair protein Ku in regulating p53 mRNA translation and function. EMBO Rep. 2016, 17, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Christian, K.J.; Lang, M.A.; Raffalli-Mathieu, F. Interaction of heterogeneous nuclear ribonucleoprotein C1/C2 with a novel cis-regulatory element within p53 mRNA as a response to cytostatic drug treatment. Mol. Pharmacol. 2008, 73, 1558–1567. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.Y.; Kim, D.Y.; Kim, S.H.; Kim, H.J.; Ryu, H.G.; Lee, J.; Lee, K.H.; Kim, K.T. Heterogeneous nuclear ribonucleoprotein (hnRNP) L promotes DNA damage-induced cell apoptosis by enhancing the translation of p53. Oncotarget 2017, 8, 51108–51122. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Kim, W.; Lee, K.H.; Kim, S.H.; Lee, H.R.; Kim, H.J.; Jung, Y.; Choi, J.H.; Kim, K.T. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ. 2013, 20, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Benchimol, S. Participation of the human p53 3′UTR in translational repression and activation following gamma-irradiation. EMBO J. 1997, 16, 4117–4125. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Ma, W.; Benchimol, S. A translation repressor element resides in the 3′ untranslated region of human p53 mRNA. Oncogene 1999, 18, 6419–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Panda, A.C.; Kang, M.J.; Guo, R.; Kim, J.; Grammatikakis, I.; Yoon, J.H.; Dudekula, D.B.; Noh, J.H.; Yang, X.; et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014, 42, 10099–10111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, D.; Goyal, A.; Ray, P.S. Interplay between RNA-binding protein HuR and microRNA-125b regulates p53 mRNA translation in response to genotoxic stress. RNA Biol. 2016, 13, 1152–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoch, A.; George, B.; Iyyappan, A.; Khan, D.; Das, S. Interplay between PTB and miR-1285 at the p53 3′UTR modulates the levels of p53 and its isoform Delta40p53alpha. Nucleic Acids Res. 2017, 45, 10206–10217. [Google Scholar] [CrossRef] [Green Version]
- Devany, E.; Zhang, X.; Park, J.Y.; Tian, B.; Kleiman, F.E. Positive and negative feedback loops in the p53 and mRNA 3′ processing pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 3351–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, R.; Ray, P.S.; Das, S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle 2008, 7, 2189–2198. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, M.; Candeias, M.M.; Malbert-Colas, L.; Mazars, A.; Fujita, J.; Olivares-Illana, V.; Fahraeus, R. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 2012, 21, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Malbert-Colas, L.; Ponnuswamy, A.; Olivares-Illana, V.; Tournillon, A.S.; Naski, N.; Fahraeus, R. HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol. Cell 2014, 54, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Munoz, M.D.; Kiselev, V.Y.; Le Novere, N.; Curk, T.; Ule, J.; Turner, M. Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat. Commun. 2017, 8, 530. [Google Scholar] [CrossRef]
- Lin, C.C.; Liao, W.T.; Yang, T.Y.; Lu, H.J.; Hsu, S.L.; Wu, C.C. MicroRNA10b modulates cisplatin tolerance by targeting p53 directly in lung cancer cells. Oncol. Rep. 2021, 46, 167. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Sfaxi, R.; Saha, A.; Monchaud, D.; Teulade-Fichou, M.P.; Vagner, S. The G-Quadruplex-Specific RNA Helicase DHX36 Regulates p53 Pre-mRNA 3′-End Processing Following UV-Induced DNA Damage. J. Mol. Biol. 2017, 429, 3121–3131. [Google Scholar] [CrossRef]
- Decorsiere, A.; Cayrel, A.; Vagner, S.; Millevoi, S. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3′-end processing and function during DNA damage. Genes Dev. 2011, 25, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Mitschka, S.; Mayr, C. Endogenous p53 expression in human and mouse is not regulated by its 3′UTR. eLife 2021, 10, e65700. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Naski, N.; Gajjar, M.; Bourougaa, K.; Malbert-Colas, L.; Fahraeus, R.; Candeias, M.M. The p53 mRNA-Mdm2 interaction. Cell Cycle 2009, 8, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Medina, I.; Garcia-Beltran, P.; de la Mora, I.; Oria-Hernandez, J.; Millot, G.; Fahraeus, R.; Reyes-Vivas, H.; Sampedro, J.G.; Olivares-Illana, V. Allosteric Interactions by p53 mRNA Govern HDM2 E3 Ubiquitin Ligase Specificity under Different Conditions. Mol. Cell Biol. 2016, 36, 2195–2205. [Google Scholar] [CrossRef] [Green Version]
- Pereg, Y.; Shkedy, D.; de Graaf, P.; Meulmeester, E.; Edelson-Averbukh, M.; Salek, M.; Biton, S.; Teunisse, A.F.; Lehmann, W.D.; Jochemsen, A.G.; et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc. Natl. Acad. Sci. USA 2005, 102, 5056–5061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakostis, K.; Ponnuswamy, A.; Fusee, L.T.; Bailly, X.; Laguerre, L.; Worall, E.; Vojtesek, B.; Nylander, K.; Fahraeus, R. p53 mRNA and p53 Protein Structures Have Evolved Independently to Interact with MDM2. Mol. Biol. Evol. 2016, 33, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakostis, K.; Fahraeus, R. Shaping the regulation of the p53 mRNA tumour suppressor: The co-evolution of genetic signatures. BMC Cancer 2019, 19, 915. [Google Scholar] [CrossRef] [Green Version]
- Karakostis, K.; Vadivel Gnanasundram, S.; Lopez, I.; Thermou, A.; Wang, L.; Nylander, K.; Olivares-Illana, V.; Fahraeus, R. A single synonymous mutation determines the phosphorylation and stability of the nascent protein. J. Mol. Cell Biol. 2019, 11, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Gueret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Vagner, S. DNA-Damage Response RNA-Binding Proteins (DDRBPs): Perspectives from a New Class of Proteins and Their RNA Targets. J. Mol. Biol. 2017, 429, 3139–3145. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, Y.; Zhang, T.; Li, L.; Chen, S.; Wu, X.; Li, H.; Qi, B.; Chen, Z. Phosphorylation of Ago2 is required for its role in DNA double-strand break repair. J. Genet. Genom. 2021, 48, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Cheng, Z.; Zhu, Q. AGO2 and its partners: A silencing complex, a chromatin modulator, and new features. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Krell, J.; Stebbing, J.; Carissimi, C.; Dabrowska, A.F.; de Giorgio, A.; Frampton, A.E.; Harding, V.; Fulci, V.; Macino, G.; Colombo, T.; et al. TP53 regulates miRNA association with AGO2 to remodel the miRNA-mRNA interaction network. Genome Res. 2016, 2016. 26, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, T.; Monteiro, A.N.A.; August, A.; Aaronson, S.A.; Hanafusa, H. BRCA1 regulates p53-dependent gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 2302–2306. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Huang, J.; Chen, J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.S.; Rajendran, P.; Dashwood, R.H. CCAR1 and CCAR2 as gene chameleons with antagonistic duality: Preclinical, human translational, and mechanistic basis. Cancer Sci. 2020, 111, 3416–3425. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Buscemi, G.; Zannini, L. Cell cycle and apoptosis regulator 2 at the interface between DNA damage response and cell physiology. Mutat. Res. Rev. Mutat. Res. 2018, 776, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nicol, S.M.; Bray, S.E.; Black, H.D.; Lorimore, S.A.; Wright, E.G.; Lane, D.P.; Meek, D.W.; Coates, P.J.; Fuller-Pace, F.V. The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell-cycle arrest after DNA damage. Oncogene 2013, 32, 3461–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Mersaoui, S.Y.; Guitton-Sert, L.; Coulombe, Y.; Song, J.; Masson, J.Y.; Richard, S. DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions. NAR Cancer 2020, 2, zcaa028. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Harris, B.R.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Deregulation of Internal Ribosome Entry Site-Mediated p53 Translation in Cancer Cells with Defective p53 Response to DNA Damage. Mol. Cell Biol. 2015, 35, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Jiang, J.; Ma, J.; Dai, S.; Xu, T.; Li, H.; Yasui, A. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS ONE 2013, 8, e60208. [Google Scholar] [CrossRef]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Abdelmohsen, K.; Gorospe, M. Regulation of HuR by DNA Damage Response Kinases. J. Nucleic Acids 2010, 2010, 981487. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, E.; Sadoughi, F.; Younesi, S.; Karimian, A.; Asemi, Z.; Farsad-Akhtar, N.; Jahanbakhshi, F.; Jamilian, H.; Yousefi, B. The molecular mechanism of nuclear signaling for degradation of cytoplasmic DNA: Importance in DNA damage response and cancer. DNA Repair 2021, 103, 103115. [Google Scholar] [CrossRef]
- Chaplin, A.K.; Hardwick, S.W.; Liang, S.; Kefala Stavridi, A.; Hnizda, A.; Cooper, L.R.; De Oliveira, T.M.; Chirgadze, D.Y.; Blundell, T.L. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat. Struct. Mol. Biol. 2021, 28, 13–19. [Google Scholar] [CrossRef]
- Kragelund, B.B.; Weterings, E.; Hartmann-Petersen, R.; Keijzers, G. The Ku70/80 ring in Non-Homologous End-Joining: Easy to slip on, hard to remove. Front. Biosci. 2016, 21, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef]
- Haronikova, L.; Vojtesek, B. HDM2 and HDMX Proteins in Human Cancer. Klin. Onkol. 2018, 31, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Seok, K.O.; Kim, Y.J.; Bae, W.K.; Lee, S.; Park, J.H. Involvement of GLTSCR2 in the DNA Damage Response. Am. J. Pathol. 2011, 179, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cho, Y.E.; Kim, Y.J.; Park, J.H. c-Jun N-terminal kinase regulates the nucleoplasmic translocation and stability of nucleolar GLTSCR2 protein. Biochem. Biophys. Res. Commun. 2016, 472, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.Y.; Kim, Y.J.; Seok, K.O.; Kim, J.H.; Chang, Y.J.; Kang, H.Y.; Park, J.H. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 2012, 19, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Chen, X. MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G(2)-M. Mol. Cell Biol. 2000, 20, 5602–5618. [Google Scholar] [CrossRef] [Green Version]
- Scoumanne, A.; Cho, S.J.; Zhang, J.; Chen, X. The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res. 2011, 39, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, E.; Zhang, J.; Chen, X. MDM2 expression is repressed by the RNA-binding protein RNPC1 via mRNA stability. Oncogene 2013, 32, 2169–2178. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.H.; Kim, R.H.; Kang, M.K.; Kim, R.H.; Kim, S.G.; Lim, P.K.; Yochim, J.M.; Baluda, M.A.; Park, N.H. p53 promotes the fidelity of DNA end-joining activity by, in part, enhancing the expression of heterogeneous nuclear ribonucleoprotein G. DNA Repair 2007, 6, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Zhao, Y.; He, H.; Sun, Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 2011, 30, 1798–1811. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tan, M.; Liu, X.; Xiong, X.; Sun, Y. Inactivation of ribosomal protein S27-like confers radiosensitivity via the Mdm2-p53 and Mdm2-MRN-ATM axes. Cell Death Dis. 2018, 9, 145. [Google Scholar] [CrossRef]
- Shah, A.; Lindquist, J.A.; Rosendahl, L.; Schmitz, I.; Mertens, P.R. Novel Insights into YB-1 Signaling and Cell Death Decisions. Cancers 2021, 13, 3306. [Google Scholar] [CrossRef]
- Sangermano, F.; Delicato, A.; Calabro, V. Y box binding protein 1 (YB-1) oncoprotein at the hub of DNA proliferation, damage and cancer progression. Biochimie 2020, 179, 205–216. [Google Scholar] [CrossRef]
- Dutertre, M.; Sanchez, G.; De Cian, M.C.; Barbier, J.; Dardenne, E.; Gratadou, L.; Dujardin, G.; Le Jossic-Corcos, C.; Corcos, L.; Auboeuf, D.; et al. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol 2010, 17, 1358–1366. [Google Scholar] [CrossRef]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Binding Factor | Binding Region (p53) | Function | Reference |
|---|---|---|---|
| Nucleolin | 5′ UTR | suppresses p53 translation | [52] |
| Rpl26 | 5′ UTR | enhances p53 translation | [52] |
| Pdcd4 | 5′ UTR | inhibits p53 translation; during DNA damage, the Pdcd4 levels are reduced, and p53 translation suppression is abrogated | [62] |
| TCP80 | 5′ UTR (IRES I) | stimulates p53 translation | [65] |
| Ku÷(Ku70/Ku80) | 5′ UTR (IRES I) | represses p53 translation; during÷DNA damage, the Ku protein is modified and abrogates binding to p53 mRNA and inhibits repression | [66] |
| hnRNP C1/C2 | 5′ UTR | stimulates p53 translation | [67] |
| hnRNP L | 5′ UTR | stimulates p53 translation | [68] |
| hnRNP Q | 5′ UTR | stimulates p53 translation | [69] |
| Dap5 | 5′ UTR (IRES I) and coding sequence (IRES II) | promotes IRES driven translation | [64] |
| PTB | 5′ UTR (IRES I), coding sequence (IRES II) and 3′UTR | regulates p53 translation | [75,77] |
| RBM38/RNPC1 | 5′ UTR and/3′ UTR | represses p53 translation | [63] |
| MDM2 | Coding sequence (IRES II) | enhances p53 translation | [44,78] |
| MDMX÷(MDM4) | Coding sequence (IRES II) | chaperoning p53 mRNA to facilitate MDM2 binding and enhances p53 translation | [79] |
| HuR | AU-rich element (3′ UTR) | stabilizes p53 mRNA and enhances translation. | [72] |
| miRNA-125b | AU-rich element (3′ UTR) | competes with HuR for binding to p53 mRNA and suppresses translation | [74] |
| PARN | AU-rich element (3′ UTR) | destabilizes p53 mRNA and decreases p53 levels under normal conditions | [76] |
| Tia1 | U-rich element (3′ UTR) | targets p53 mRNA to stress granules under normal conditions; during DNA damage, enhances p53 translation | [80] |
| 40 kDa unknown protein | U-rich element (3′ UTR) | negatively regulates p53 levels under normal conditions, which were relieved during DNA damage | [70] |
| miR-10b | 3′ UTR | regulates the stability of p53 mRNA during cisplatin treatment | [81] |
| RBP | Main Feature | Functional Interplay with p53 | Reference |
|---|---|---|---|
| AGO2 | RNA interference | p53 regulates AGO2 association with miRNAs and remodel miRNA-mRNA network during DDR | [96,97,98] |
| BRCA1 | E3 ubiquitin-protein ligase | DNA damage sensor; regulates p53 dependent gene expression | [99,100,101,102] |
| CCAR2 | nuclear protein | activates p53 and induction of p53 dependent apoptosis | [103,104] |
| DAP5 (EIF4G2) | translation initiation factor | stimulates p53 translation under different stress conditions | [64] |
| DDX5÷(p68) | ATP-dependent RNA helicase, transcriptional regulator | required for p53 dependent p21 induction and cell cycle arrest | [105,106] |
| DHX9 (RHA) | ATP-dependent RNA helicase | enhances expression of p53 | [65,107] |
| hnRNP F/H | nuclear ribonucleoprotein | essential for maintaining p53 pre-mRNA 3′-end processing | [83] |
| hnRNPC 1/2 | nuclear ribonucleoprotein | interacts with p53 5′ UTR and stimulates translation | [67] |
| hnRNPUL-1 | transcription regulator | interacts with p53 and inhibits its transcriptional activity during DDR | [108,109] |
| HuR | RBP; interacts with 3′ UTRs of mRNAs | increases the stability of p53 mRNA and translation | [72,110,111] |
| Ku70-Ku80÷(XCCR5-XCCR6) | single-stranded DNA-dependent ATP-dependent helicases; DNA damage sensor | interacts with p53 5′ UTR and suppresses translation under normal conditions; during DDR, Ku protein is modified and releases the suppression of p53 translation | [112,113,114] |
| MDM2 | E3 ubiquitin ligase | enhances p53 translation during DDR | [26,78,87,115] |
| NOP53 | nuclear protein | DNA damage sensor, essential for stabilization of p53 protein | [116,117,118] |
| Nucleolin | nucleolar protein; multifunctional phosphoprotein | represses p53 translation | [53,54] |
| PCBP4÷(MCG10) | poly(C)-binding protein | p53 activates PCBP4 during DDR and induces apoptosis and cell cycle arrest | [119,120] |
| PDCD4 | apoptosis regulation protein | suppresses p53 in normal condition, under DDR PDCD4 levels are reduced | [62] |
| RBM38÷(RNPC1) | RBP; regulates mRNA stability | represses MDM2 and p53 translation | [63,121] |
| RBMX÷(hnRNP G) | nuclear ribonucleoprotein | p53 enhances the expression of RBMX and promotes DNA repair | [122,123] |
| RPL26 | 60S ribosomal protein | enhances p53 translation after DNA damage | [52,53,54] |
| RPS27L | ribosomal protein | direct p53-inducible target, interferes with p53-MDM2 axis | [124,125] |
| TCP80 (IL3) | regulates p53 IRES translation | interacts with 5′ UTR and stimulates p53 translation | [65,107] |
| YB-1 and EWS | multifunctional nucleic acid binding proteins | regulate MDM2 splicing and increase p53 levels during DDR | [126,127,128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadivel Gnanasundram, S.; Bonczek, O.; Wang, L.; Chen, S.; Fahraeus, R. p53 mRNA Metabolism Links with the DNA Damage Response. Genes 2021, 12, 1446. https://doi.org/10.3390/genes12091446
Vadivel Gnanasundram S, Bonczek O, Wang L, Chen S, Fahraeus R. p53 mRNA Metabolism Links with the DNA Damage Response. Genes. 2021; 12(9):1446. https://doi.org/10.3390/genes12091446
Chicago/Turabian StyleVadivel Gnanasundram, Sivakumar, Ondrej Bonczek, Lixiao Wang, Sa Chen, and Robin Fahraeus. 2021. "p53 mRNA Metabolism Links with the DNA Damage Response" Genes 12, no. 9: 1446. https://doi.org/10.3390/genes12091446
APA StyleVadivel Gnanasundram, S., Bonczek, O., Wang, L., Chen, S., & Fahraeus, R. (2021). p53 mRNA Metabolism Links with the DNA Damage Response. Genes, 12(9), 1446. https://doi.org/10.3390/genes12091446