
Variation and Selection in the Putative Sperm-Binding Region of ZP3 in Muroid Rodents: A Comparison between Cricetids and Murines

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, DNA Extraction, Amplification and Sequencing
2.2. Sequence Analyses
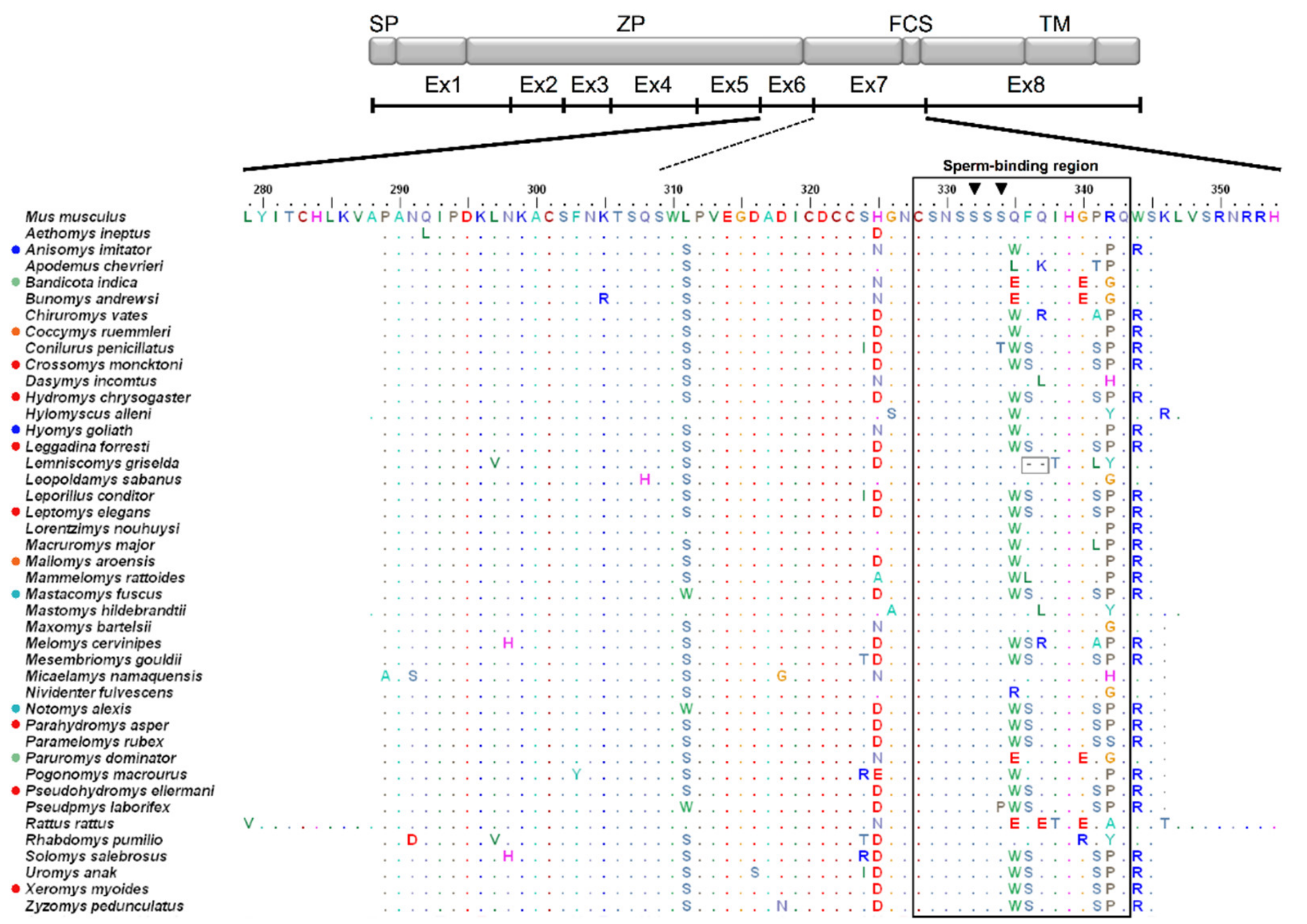
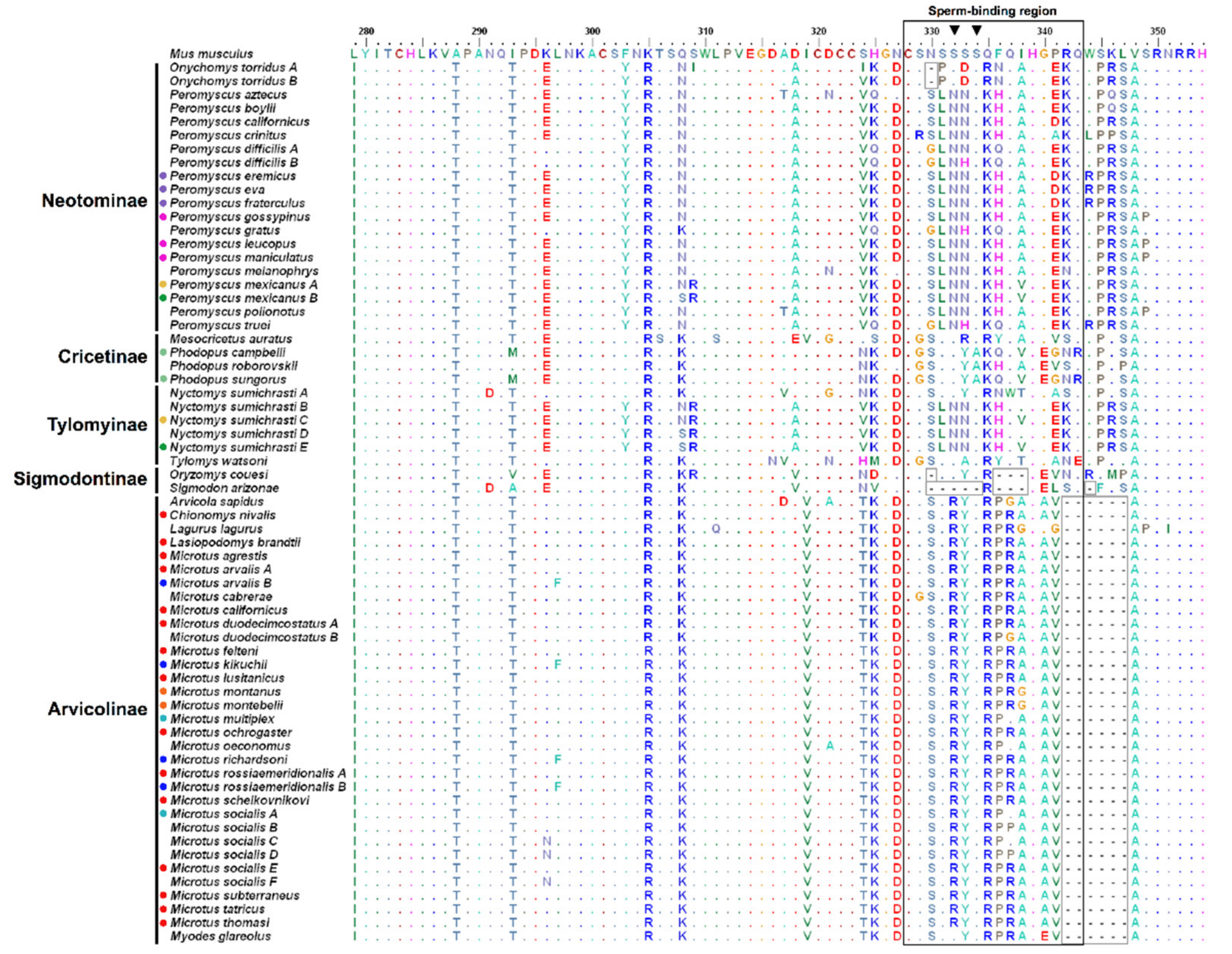
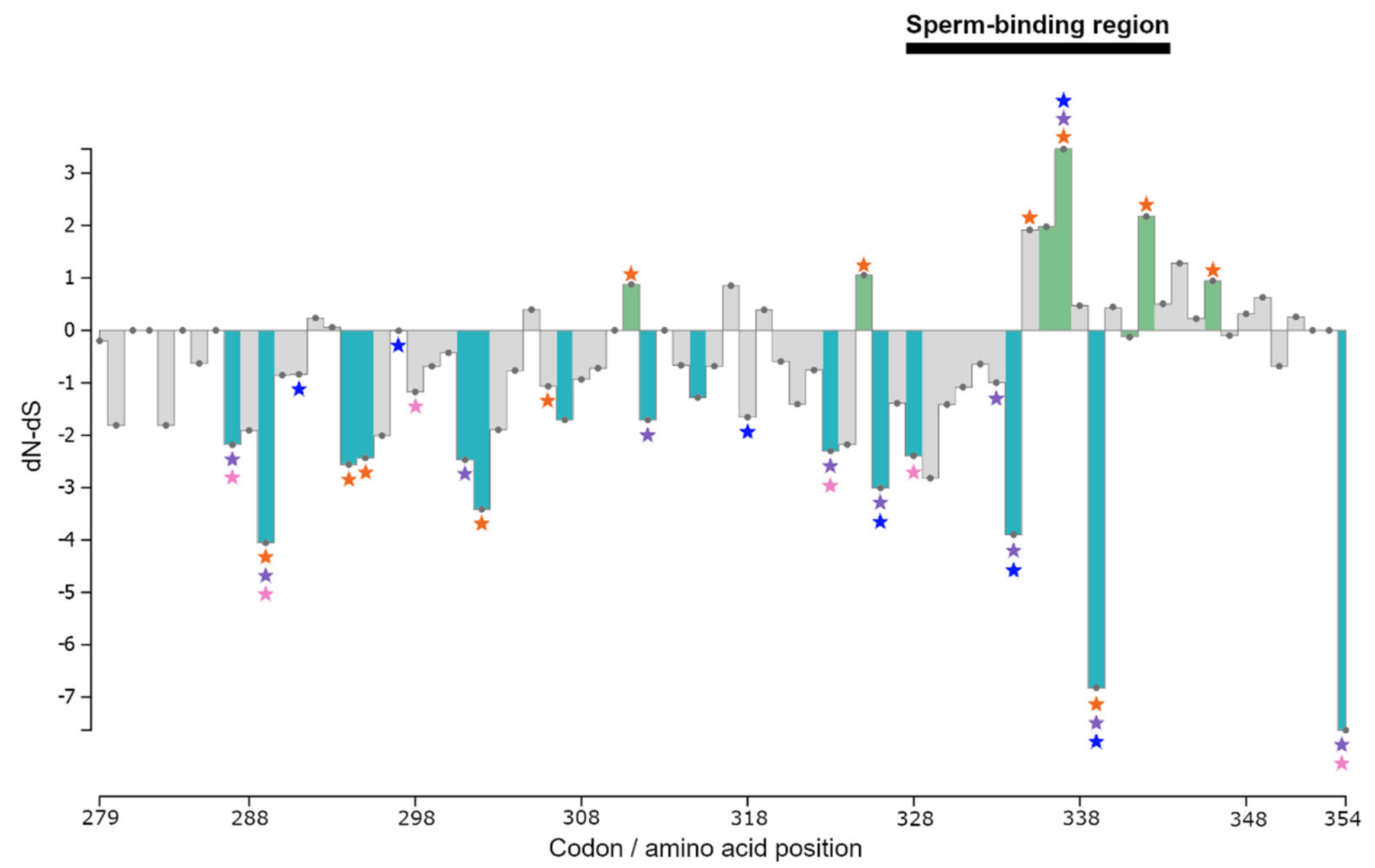
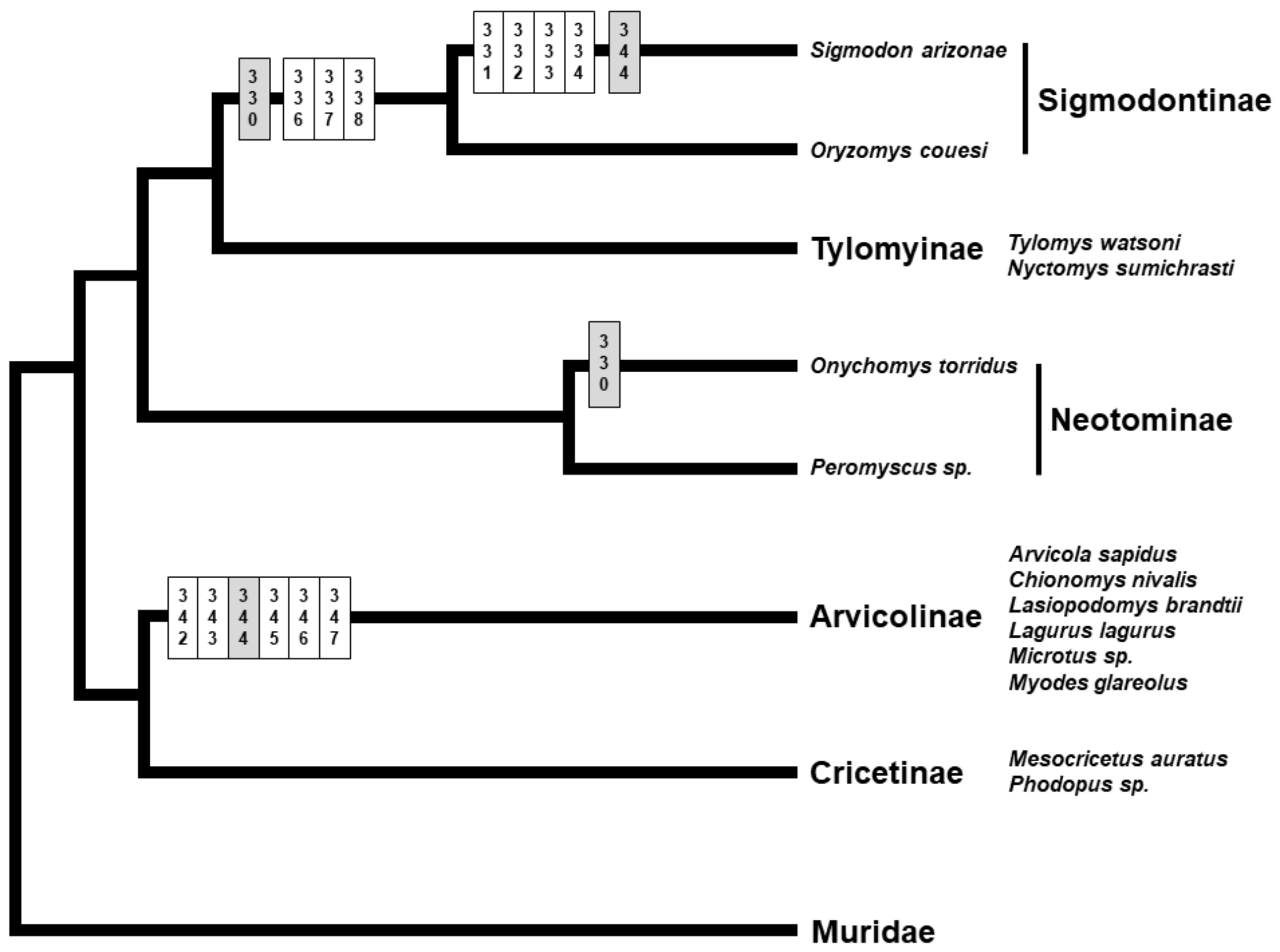
3. Results
3.1. Genetic Variation and Phylogeny
3.2. Amino Acid Variation
3.3. Selection Tests
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swanson, W.J.; Vacquier, V.D. The rapid evolution of reproductive proteins. Nat. Rev. Genet. 2002, 3, 137–144. [Google Scholar] [CrossRef]
- Turner, L.M.; Hoekstra, H.E. Causes and consequences of the evolution of reproductive proteins. Int. J. Dev. Biol. 2008, 52, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.D.; Swanson, W.J. Proteomics enhances evolutionary and functional analysis of reproductive proteins. BioEssays 2010, 32, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.-P.; Bank, C.; Brännström, A.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, N.L.; Gasper, J.; Sekino, M.; Springer, S.A.; Aquadro, C.F.; Swanson, W.J. Coevolution of interacting fertilization proteins. PLoS Genet. 2009, 5, e1000570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassarman, P.M.; Litscher, E.S. Sperm-egg recognition mechanisms in mammals. Curr. Top Dev. Biol. 1995, 30, 1–19. [Google Scholar]
- Wassarman, P.M. Mammalian fertilization: Review molecular aspects of gamete adhesion, exocytosis, and fusion. Cell 1999, 96, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Kinloch, R.A.; Wassarman, P.M. Nucleotide sequence of the gene encoding zona pellucida glycoprotein ZP3—The mouse sperm receptor. Nucleic Acids Res. 1989, 17, 2861–2863. [Google Scholar] [CrossRef] [Green Version]
- Bleil, J.D.; Wassarman, P.M. Structure and function of the zona pellucida: Identification and characterization of the proteins of the mouse oocyte’s zona pellucida. Dev. Biol. 1980, 76, 185–202. [Google Scholar] [CrossRef]
- Bleil, J.D.; Wassarman, P.M. Autoradiographic visualization of the mouse egg’s sperm receptor bound to sperm. J. Cell Biol. 1986, 102, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Wassarman, P.M.; Jovine, L.; Qi, H.; Williams, Z.; Darie, C.; Litscher, E.S. Recent aspects of mammalian fertilization research. Mol. Cell. Endocrinol. 2005, 234, 95–103. [Google Scholar] [CrossRef]
- Litscher, E.S.; Williams, Z.; Wassarman, P.M. Zona pellucida glycoprotein ZP3 and fertilization in mammals. Mol. Reprod. Dev. 2009, 76, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Florman, H.M.; Wassarman, P.M. O-linked oligosaccharides of mouse egg ZP3 account for its sperm receptor activity. Cell 1985, 41, 313–324. [Google Scholar] [CrossRef]
- Rosière, T.K.; Wassarman, P.M. Identification of a region of mouse zona pellucida glycoprotein mZP3 that possesses sperm receptor activity. Dev. Biol. 1992, 154, 309–317. [Google Scholar] [CrossRef]
- Kinloch, R.A.; Sakai, Y.; Wassarman, P.M. Mapping the mouse ZP3 combining site for sperm by exon swapping and site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Litscher, E.S.; Wassarman, P.M. Inactivation of the mouse sperm receptor, mZP3, by site-directed mutagenesis of individual serine residues located at the combining site for sperm. Proc. Natl. Acad. Sci. USA 1998, 95, 6193–6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redgrove, K.A.; Aitken, R.J.; Nixon, B. More than a simple lock and key mechanism: Unraveling the intricacies of sperm-zona pellucida binding. In Binding Protein; Abdelmohsen, K., Ed.; InTech: Rijeka, Croatia, 2012; pp. 73–122. [Google Scholar]
- Tumova, L.; Zigo, M.; Sutovsky, P.; Sedmikova, M.; Postlerova, P. Ligands and Receptors Involved in the Sperm-Zona Pellucida Interactions in Mammals. Cells 2021, 10, 133. [Google Scholar] [CrossRef]
- Chalabi, S.; Panico, M.; Sutton-Smith, M.; Haslam, S.M.; Patankar, M.S.; Lattanzio, F.A.; Morris, H.R.; Clarck, G.F.; Dell, A. Differential O-glycosylation of a conserved domain expressed in murine and human ZP3. Biochem 2006, 45, 637–647. [Google Scholar] [CrossRef]
- Monné, M.; Jovine, L. A structural view of egg coat architecture and function in fertilization. Biol. Reprod. 2011, 85, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Rankin, T.L.; Tong, Z.B.; Castle, P.E.; Lee, E.; Gore-Langton, R.; Nelson, L.M.; Dean, J. Human ZP3 restores fertility in Zp3 null mice without affecting order-specific sperm binding. Development 1998, 125, 2415–2424. [Google Scholar] [CrossRef]
- Dean, J. Reassessing the molecular biology of sperm-egg recognition with mouse genetics. Bioessays 2004, 26, 29–38. [Google Scholar] [CrossRef]
- Clark, G.F.; Dell, A. Molecular models for murine sperm-egg binding. J. Biol. Chem. 2006, 281, 13853–13856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahlay, G.; Gauthier, L.; Baibakov, B.; Epifano, O.; Dean, J. Gamete recognition in mice depends on the cleavage status of an egg’s zona pellucida protein. Science 2010, 329, 216–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avella, M.A.; Baibakov, B.; Dean, J. A single domain of the ZP2 zona pellucida protein mediates gamete recognition in mice and humans. J. Cell Biol. 2014, 205, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, E.; Wright, G.J. Find and fuse: Unsolved mysteries in sperm–egg recognition. PLoS Biol. 2020, 18, e3000953. [Google Scholar] [CrossRef] [PubMed]
- Stsiapanava, A.; Xu, C.; Brunati, M.; Zamora-Caballero, S.; Schaeffer, C.; Bokhove, M.; Han, L.; Hebert, H.; Carroni, M.; Yasumasu, S.; et al. Cryo-EM structure of native human uromodulin, a zona pellucida module polymer. EMBO J. 2020, 39, e106807. [Google Scholar] [CrossRef]
- Swann, C.A.; Cooper, S.J.B.; Breed, W.G. Molecular evolution of the carboxy terminal region of the zona pellucida 3 glycoprotein in murine rodents. Reproduction 2007, 133, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, W.J.; Yang, Z.; Wolfner, M.F.; Aquadro, C.F. Positive Darwinian selection drives the evolution of several female reproductive proteins in mammals. Proc. Natl. Acad. Sci. USA 2001, 98, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.C.; Loughran, N.B.; Walsh, T.A.; Harrison, A.J.; O’Connell, M.J. Positive selection neighboring functionally essential sites and disease-implicated regions of mammalian reproductive proteins. BMC Evol. Biol. 2010, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Swann, C.C.; Hart, M.W. Molecular evolution of mammalian genes with epistatic interactions in fertilization. BMC Evol. Biol. 2019, 19, 154. [Google Scholar]
- Jansa, S.A.; Lundrigan, B.L.; Tucker, P.K. Tests for positive selection on immune and reproductive genes in closely related species of the murine genus. Mus. J. Mol. Evol. 2003, 56, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, L.M.; Hoekstra, H.E. Adaptive evolution of fertilization proteins within a genus: Variation in ZP2 and ZP3 in deer mice (Peromyscus). Mol. Biol. Evol. 2006, 23, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, L.M.; Hoekstra, H.E. Reproductive protein evolution within and between species: Maintenance of divergent ZP3 alleles in Peromyscus. Mol. Ecol. 2008, 17, 2616–2628. [Google Scholar] [CrossRef]
- Swann, C.A.; Cooper, S.J.B.; Breed, W.G. The egg coat zona pellucida 3 glycoprotein—Evolution of its putative sperm-binding region in Old World murine rodents (Rodentia: Muridae). Reprod. Fertil. Dev. 2017, 29, 2376–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, A.R.; Möller, L.M.; Beheregaray, L.B.; Coelho, M.M. Evolution of 2 reproductive proteins, ZP3 and PKDREJ, in cetaceans. J. Hered. 2011, 102, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Costa, V.; Beja-Pereira, A. Evolutionary patterns of two major reproduction candidate genes (Zp2 and Zp3) reveal no contribution to reproductive isolation between bovine species. BMC Evol. Biol. 2011, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaline, J.; Brunet-Lecomte, P.; Montuire, S.; Viriot, L.; Courant, F. Anatomy of the arvicoline radiation (Rodentia): Palaeogeographical, palaeoecological history and evolutionary data. Ann. Zool. Fenn. 1999, 36, 239–267. [Google Scholar]
- Musser, G.M.; Carleton, M.D. Family Cricetidae. In Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D.E., Reeder, D.M., Eds.; Smithsonian Institution: Washington, DC, USA, 1993; pp. 955–1189. [Google Scholar]
- Fink, S.; Fischer, M.C.; Excoffier, L.; Heckel, G. Genomic scans support repetitive continental colonization events during the rapid radiation of voles (Rodentia: Microtus): The utility of AFLPs versus mitochondrial and nuclear sequence markers. Syst. Biol. 2010, 59, 548–572. [Google Scholar] [CrossRef] [Green Version]
- Bastos-Silveira, C.; Santos, S.M.; Monarca, R.; Mathias, M.L.; Heckel, G. Deep mitochondrial introgression and hybridization among ecologically divergent vole species. Mol. Ecol. 2012, 21, 5309–5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paupério, J.; Herman, J.S.; Melo-Ferreira, J.; Jaarola, M.; Alves, P.C.; Searle, J.B. Cryptic speciation in the field vole: A multilocus approach confirms three highly divergent lineages in Eurasia. Mol. Ecol. 2012, 21, 6015–6032. [Google Scholar] [CrossRef] [PubMed]
- Beysard, M.; Heckel, G. Structure and dynamics of hybrid zones at different stages of speciation in the common vole (Microtus arvalis). Mol. Ecol. 2014, 23, 673–687. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritschi, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Stephens, M.; Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.; Smith, N.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Curr. Protoc. Bioinform. 2003, 6, 6.5.1–6.5.14. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control. 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K. SeqState—Primer design and sequence statistics for phylogenetic DNA data sets. Appl. Bioinform. 2005, 4, 65–69. [Google Scholar]
- Simmons, M.P.; Ochoterena, H. Gaps as characters in sequence-based phylogenetic analyses. Syst. Biol. 2000, 49, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K. Incorporating information from length-mutational events into phylogenetic analysis. Mol. Phylogenet. Evol. 2006, 38, 667–676. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. 2010. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 9 March 2016).
- Arenas, M.; Posada, D. Coalescent Simulation of Intracodon Recombination. Genetics 2010, 184, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, M.; Posada, D. The influence of recombination on the estimation of selection from coding sequence alignments. In Natural Selection: Methods and Applications; Fares, M.A., Ed.; CRC Press/Taylor & Francis: London, UK, 2014; pp. 112–125. [Google Scholar]
- Del Amparo, R.; Branco, C.; Arenas, J.; Vicens, A.; Arenas, M. Analysis of selection in protein-coding sequences accounting for common biases. Brief Bioinform. 2021, 22, bbaa431. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Salminen, M. Identification of breakpoints in intergenotypic recombinants of HIV type I by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.-M.K. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Swanson, W.J.; Nielsen, R.; Yang, Q.F. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 2003, 20, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.S.W.; Yang, Z.; Goldman, N.; Nielsen, R. Accuracy and Power of Statistical Methods for Detecting Adaptive Evolution in Protein Coding Sequences and for Identifying Positively Selected Sites. Genetics 2004, 168, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–24729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; dos Reis, M. Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 2001, 28, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Berlin, S.; Smith, N.G. Testing for adaptive evolution of the female reproductive protein ZPC in mammals, birds and fishes reveals problems with the M7-M8 likelihood ratio test. BMC Evol. Biol. 2005, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.K.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Gene. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Murrell, N.; Fourment, M.; Frost, S.D.W.; Delport, W.; Scheffler, K. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, S.; Adkins, R.; Anderson, J. Phylogeny and divergence-date estimates of rapid radiations in muroid rodents based on multiple nuclear genes. Syst. Biol. 2004, 53, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Fabre, P.-H.; Hautier, L.; Dimitrov, D.; Douzery, E.J.P. A glimpse on the pattern of rodent diversification: A phylogenetic approach. BMC Evol. Biol. 2012, 12, 88. [Google Scholar] [CrossRef] [Green Version]
- Steppan, S.J.; Schenk, J.J. Muroid rodent phylogenetics: 900-species tree reveals increasing diversification rates. PLoS ONE 2017, 12, e0183070. [Google Scholar] [CrossRef] [Green Version]
- Jaarola, M.; Martínková, N.; Gündüz, I.; Brunhoff, C.; Zima, J.; Nadachowski, A.; Amori, G.; Bulatova, N.S.; Chondropoulos, B.; Fraguedakis-Tsolis, S.; et al. Molecular phylogeny of the speciose vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2004, 33, 647–663. [Google Scholar] [CrossRef]
- Martínková, N.; Moravec, J. Multilocus phylogeny of arvicoline voles (Arvicolini, Rodentia) shows small tree terrace size. Folia Zool. 2012, 61, 254–267. [Google Scholar] [CrossRef]
- Hogan, K.M.; Davis, S.K.; Greenbaum, I.F. Mitochondrial-DNA Analysis of the Systematic Relationships within the Peromyscus maniculatus Species Group. J. Mammal. 1997, 78, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Bradley, R.D.; Durish, N.D.; Rogers, D.S.; Miller, J.R.; Engstrom, M.D.; Kilpatrick, C.W. Toward a Molecular Phylogeny for Peromyscus: Evidence from Mitochondrial Cytochrome-b Sequences. J. Mammal. 2007, 88, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.N., II; Amman, B.R.; Keith, M.S.; Thompson, C.W.; Bradley, R.D. What Is Peromyscus? Evidence from nuclear and mitochondrial DNA sequences suggests the need for a new classification. J. Mammal. 2015, 96, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gering, E.J.; Opazo, J.C.; Storz, J.F. Molecular evolution of cytochrome b in high- and low-altitude deer mice (genus Peromyscus). Heredity 2009, 102, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Neumann, K.; Michaux, J.; Lebedev, V.; Yigit, N.; Colak, E.; Ivanova, N.; Poltoraus, A.; Surov, A.; Markov, G.; Maak, S.; et al. Molecular phylogeny of the Cricetinae subfamily based on the mitochondrial cytochrome b and 12S rRNA genes and the nuclear vWF gene. Mol. Phylogenet. Evol. 2006, 39, 135–148. [Google Scholar] [CrossRef]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and Power of the Likelihood Ratio Test in Detecting Adaptive Molecular Evolution. Mol. Biol. Evol. 2001, 18, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Nei, M. False-Positive Selection Identified by ML-Based Methods: Examples from the Sig1 Gene of the Diatom Thalassiosira weissflogii and the tax Gene of a Human T-cell Lymphotropic Virus. Mol. Biol. Evol. 2004, 21, 914–921. [Google Scholar] [CrossRef]
- Suzuki, Y. False-positive results obtained from the branch-site test of positive selection. Genes Genet. Syst. 2008, 83, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimova, M.; Yang, Z. Multiple Hypothesis Testing to Detect Lineages under Positive Selection that Affects Only a Few Sites. Mol. Biol. Evol. 2007, 24, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Suzuki, Y.; Nei, M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc. Natl. Acad. Sci. USA 2009, 106, 6700–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and Power of Bayes Prediction of Amino Acid Sites under Positive Selection. Mol. Biol. Evol. 2002, 19, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Visconti, P.E.; Florman, H.E. Mechanisms of sperm-egg interactions: Between sugars and broken bonds. Sci. Signal. 2010, 3, pe35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, G.F. The molecular basis of mouse sperm–zona pellucida binding: A still unresolved issue in developmental biology. Reproduction 2011, 142, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Moros-Nicolás, C.; Chevret, P.; Jiménez-Movilla, M.; Algarra, B.; Cots-Rodríguez, P.; González-Brusi, L.; Avilés, M.; Izquierdo-Rico, M.J. New Insights into the Mammalian Egg Zona Pellucida. Int. J. Mol. Sci. 2021, 22, 3276. [Google Scholar] [CrossRef] [PubMed]
- Cerveira, A.M.; Soares, J.; Bastos-Silveira, C.; Mathias, M.L. Reproductive isolation between sister species of Iberian pine voles, Microtus duodecimcostatus and M. lusitanicus. Ethol. Ecol. Evol. 2018, 31, 121–139. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | LRT | 2∆l | d.f. | p Value |
|---|---|---|---|---|
| Site-models: Cricetidae + Murinae | M0 vs. M3 | 273.646 | 4 | <0.001 |
| M1a vs. M2a | 37.492 | 2 | <0.001 | |
| M7 vs. M8 | 42.397 | 2 | <0.001 | |
| M8a vs. M8 | 35.522 | 1 | <0.001 | |
| Site-models: Cricetidae | M0 vs. M3 | 139.759 | 4 | <0.001 |
| M1a vs. M2a | 0.726 | 2 | 0.696 | |
| M7 vs. M8 | 4.4671 | 2 | 0.107 | |
| M8a vs. M8 | 3.490 | 1 | 0.062 | |
| Site-models: Murinae | M0 vs. M3 | 102.930 | 4 | <0.001 |
| M1a vs. M2a | 16.674 | 2 | <0.001 | |
| M7 vs. M8 | 21.164 | 2 | <0.001 | |
| M8a vs. M8 | 18.286 | 1 | <0.001 | |
| Site-models: Microtus | M0 vs. M3 | 60.304 | 4 | <0.001 |
| M1a vs. M2a | 8.622 | 2 | 0.013 | |
| M7 vs. M8 | 8.350 | 2 | 0.015 | |
| M8a vs. M8 | 8.232 | 1 | 0.004 | |
| Site-models: Peromyscus | M0 vs. M3 | 15.034 | 4 | 0.004 |
| M1a vs. M2a | 0.000 | 2 | 1.000 | |
| M7 vs. M8 | 0.025 | 2 | 0.988 | |
| M8a vs. M8 | 0.022 | 1 | 0.883 | |
| Branch-site models: Murinae | null vs. MA1 | 0.796 | 2 | <0.001 |
| M1a vs. MA1 | 1.420 | 2 | <0.001 | |
| Branch-site models: Cricetidae | null vs. MA1 | 1.077 | 2 | <0.001 |
| M1a vs. MA1 | 12.208 | 2 | <0.001 | |
| Branch-site models: Arvicolinae | null vs. MA1 | 6.280 | 2 | <0.001 |
| M1a vs. MA1 | 6.560 | 2 | <0.001 | |
| Branch-site models: Cricetinae | null vs. MA1 | 0.000 | 2 | 1.000 |
| M1a vs. MA1 | 0.927 | 2 | 0.629 | |
| Branch-site models: Neotominae | null vs. MA1 | 0.000 | 2 | 1.000 |
| M1a vs. MA1 | 0.000 | 2 | 1.000 | |
| Branch-site models: Sigmodontinae | null vs. MA1 | 0.000 | 2 | 1.000 |
| M1a vs. MA1 | 0.004 | 2 | 0.998 | |
| Branch-site models: Tylomyinae | null vs. MA1 | 0.087 | 2 | <0.001 |
| M1a vs. MA1 | 0.242 | 2 | 0.242 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, M.A.; Fernandes, C.R.; Heckel, G.; da Luz Mathias, M.; Bastos-Silveira, C. Variation and Selection in the Putative Sperm-Binding Region of ZP3 in Muroid Rodents: A Comparison between Cricetids and Murines. Genes 2021, 12, 1450. https://doi.org/10.3390/genes12091450
Duarte MA, Fernandes CR, Heckel G, da Luz Mathias M, Bastos-Silveira C. Variation and Selection in the Putative Sperm-Binding Region of ZP3 in Muroid Rodents: A Comparison between Cricetids and Murines. Genes. 2021; 12(9):1450. https://doi.org/10.3390/genes12091450
Chicago/Turabian StyleDuarte, Margarida Alexandra, Carlos Rodríguez Fernandes, Gerald Heckel, Maria da Luz Mathias, and Cristiane Bastos-Silveira. 2021. "Variation and Selection in the Putative Sperm-Binding Region of ZP3 in Muroid Rodents: A Comparison between Cricetids and Murines" Genes 12, no. 9: 1450. https://doi.org/10.3390/genes12091450
APA StyleDuarte, M. A., Fernandes, C. R., Heckel, G., da Luz Mathias, M., & Bastos-Silveira, C. (2021). Variation and Selection in the Putative Sperm-Binding Region of ZP3 in Muroid Rodents: A Comparison between Cricetids and Murines. Genes, 12(9), 1450. https://doi.org/10.3390/genes12091450