
Novel Approach Combining Transcriptional and Evolutionary Signatures to Identify New Multiciliation Genes

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Functional Genomics Analysis
2.1.1. Public Transcriptomic Datasets
2.1.2. Raw Data Analysis
2.1.3. Overexpressed Genes Clustering
2.2. Comparative Genomics Analysis
2.2.1. Evolutionary Analysis of Multiciliated Genes
2.2.2. Search for Atypically Conserved Genes in Otomorpha
2.3. In Depth Analysis of Target Genes
2.3.1. Generation of Interaction Networks
2.3.2. Computation and Clustering of Phylogenetic Profiles
3. Results
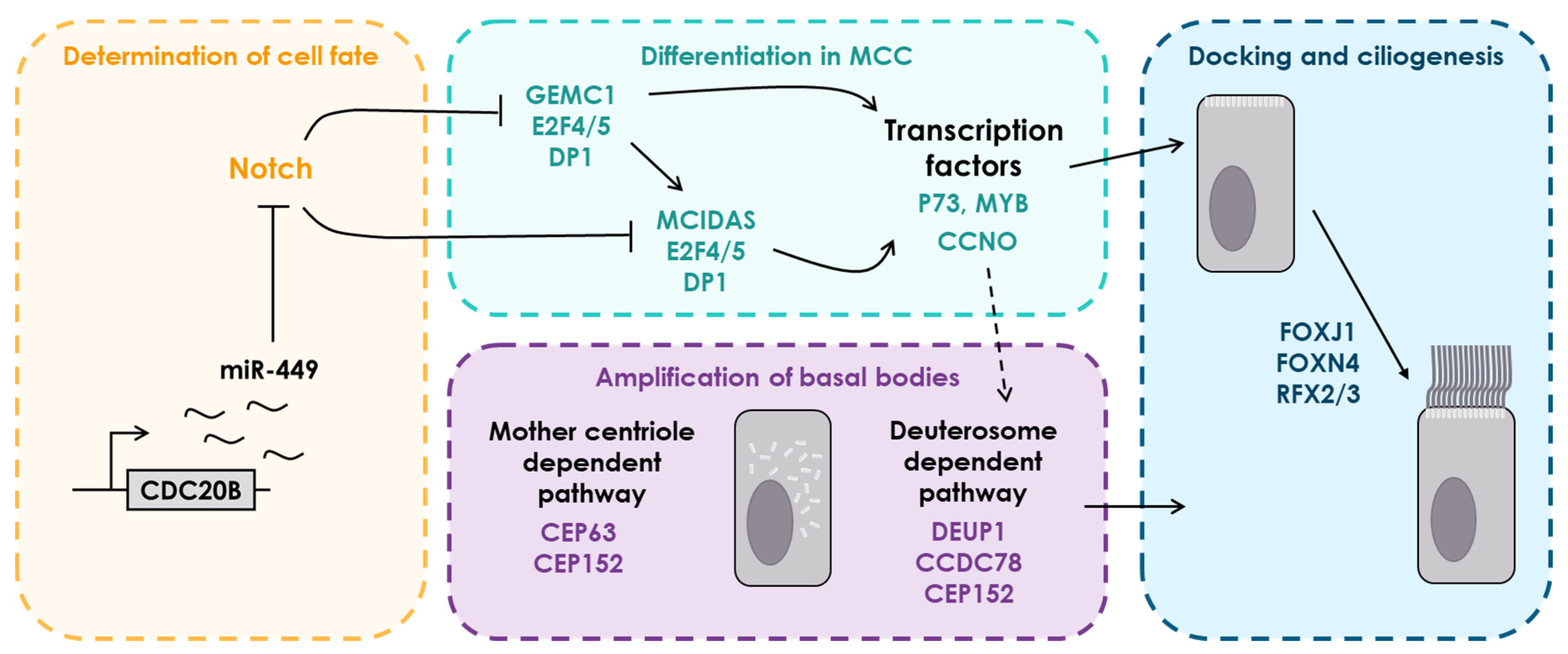
3.1. Functional Analysis of Multiciliation Oriented Experiments
3.1.1. Gene Overexpression
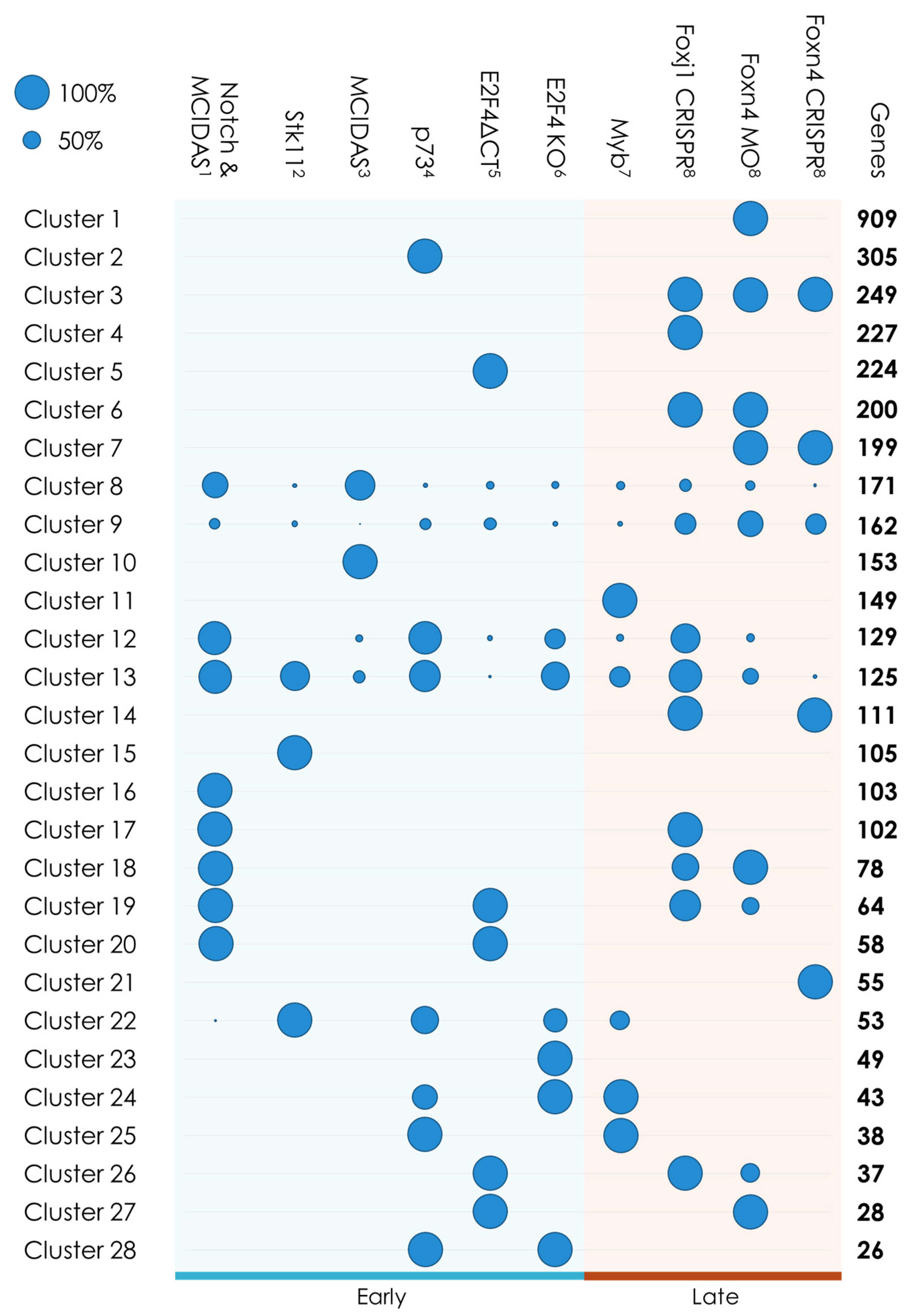
3.1.2. Gene Clustering
3.2. Comparative Genomics
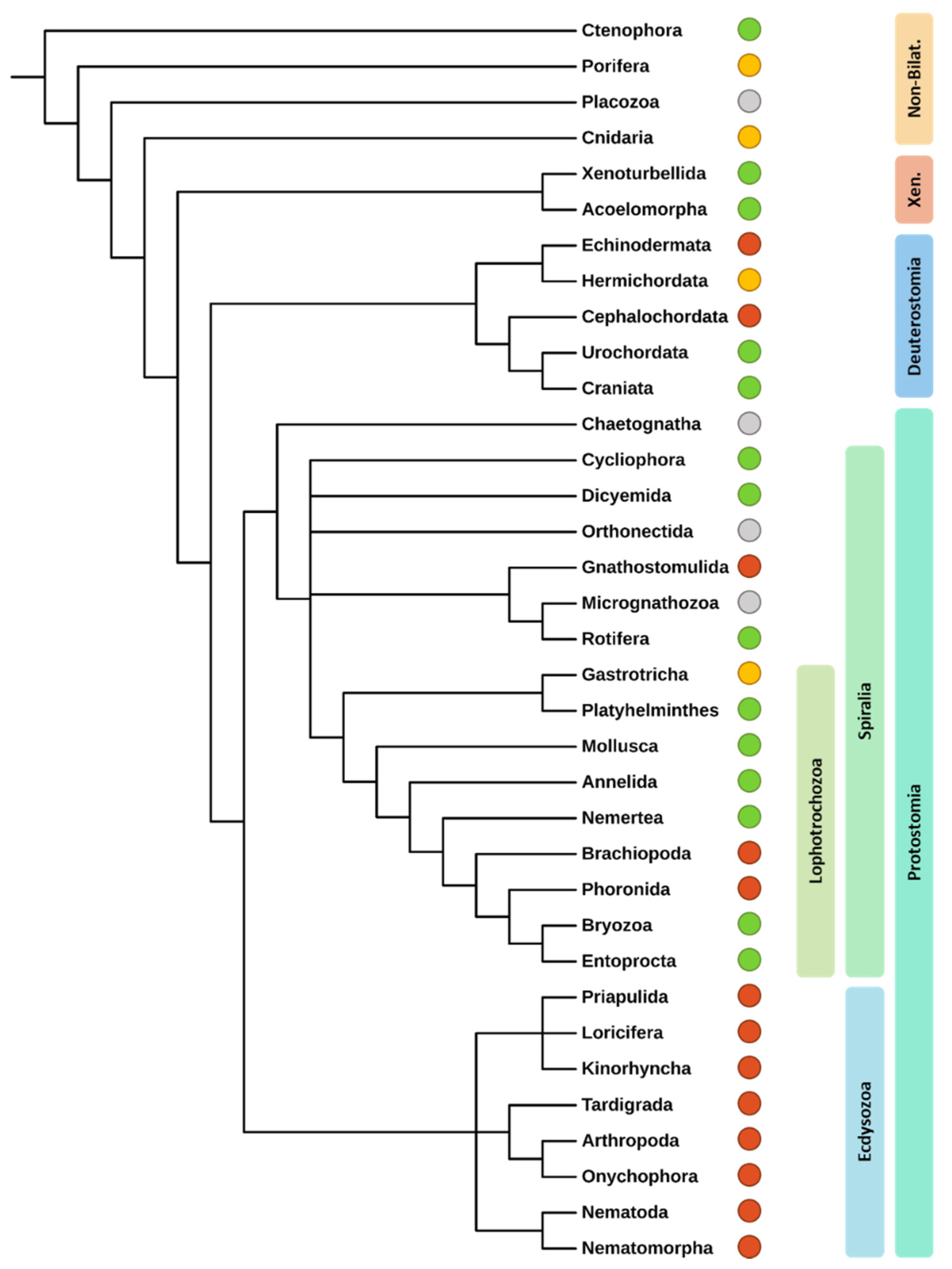
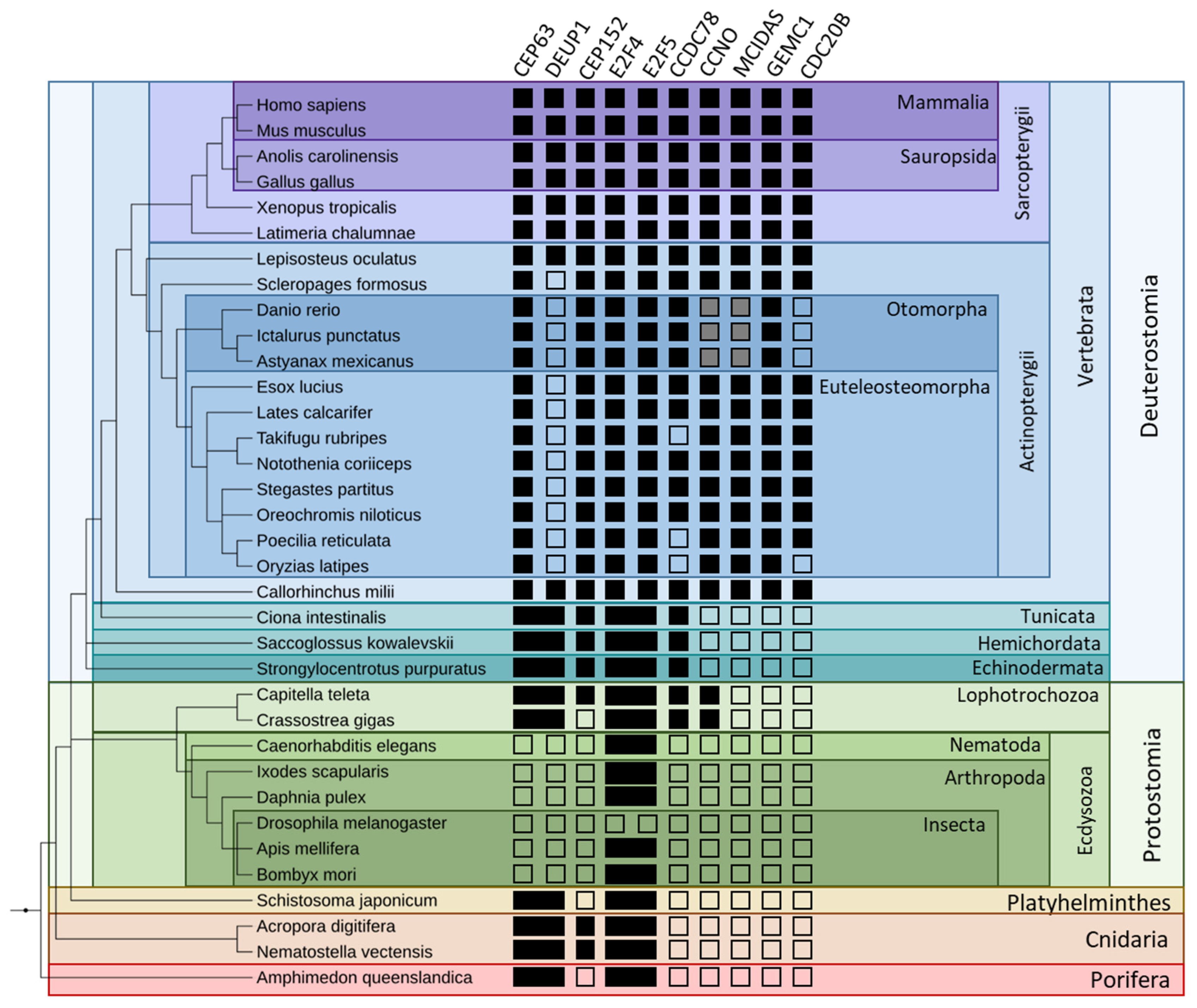
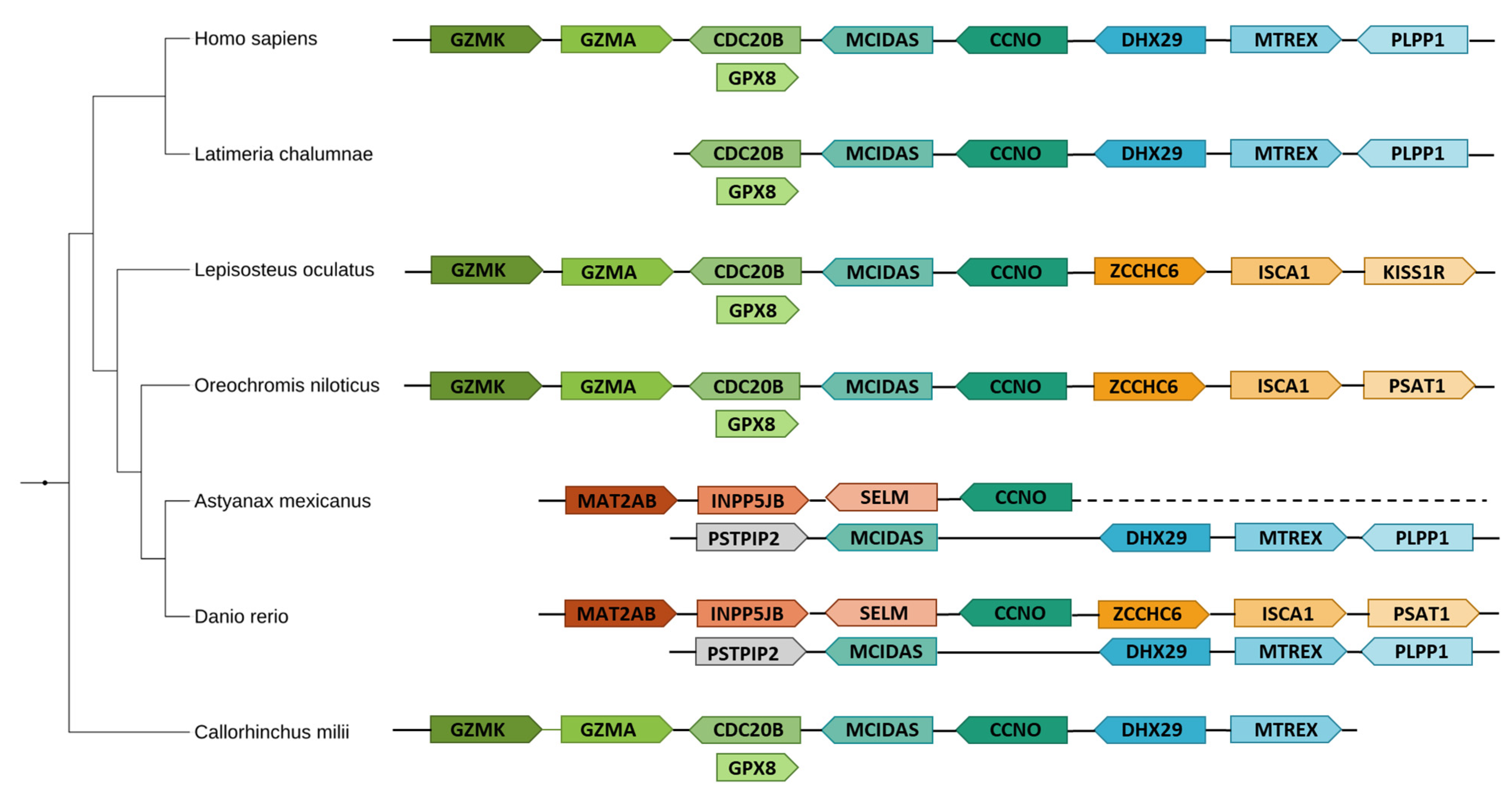
3.2.1. Identification of Evolutionary Pattern
3.2.2. Multi-Level Identification of Differential Conservation
3.3. Integration of Functional and Comparative Genomics Results
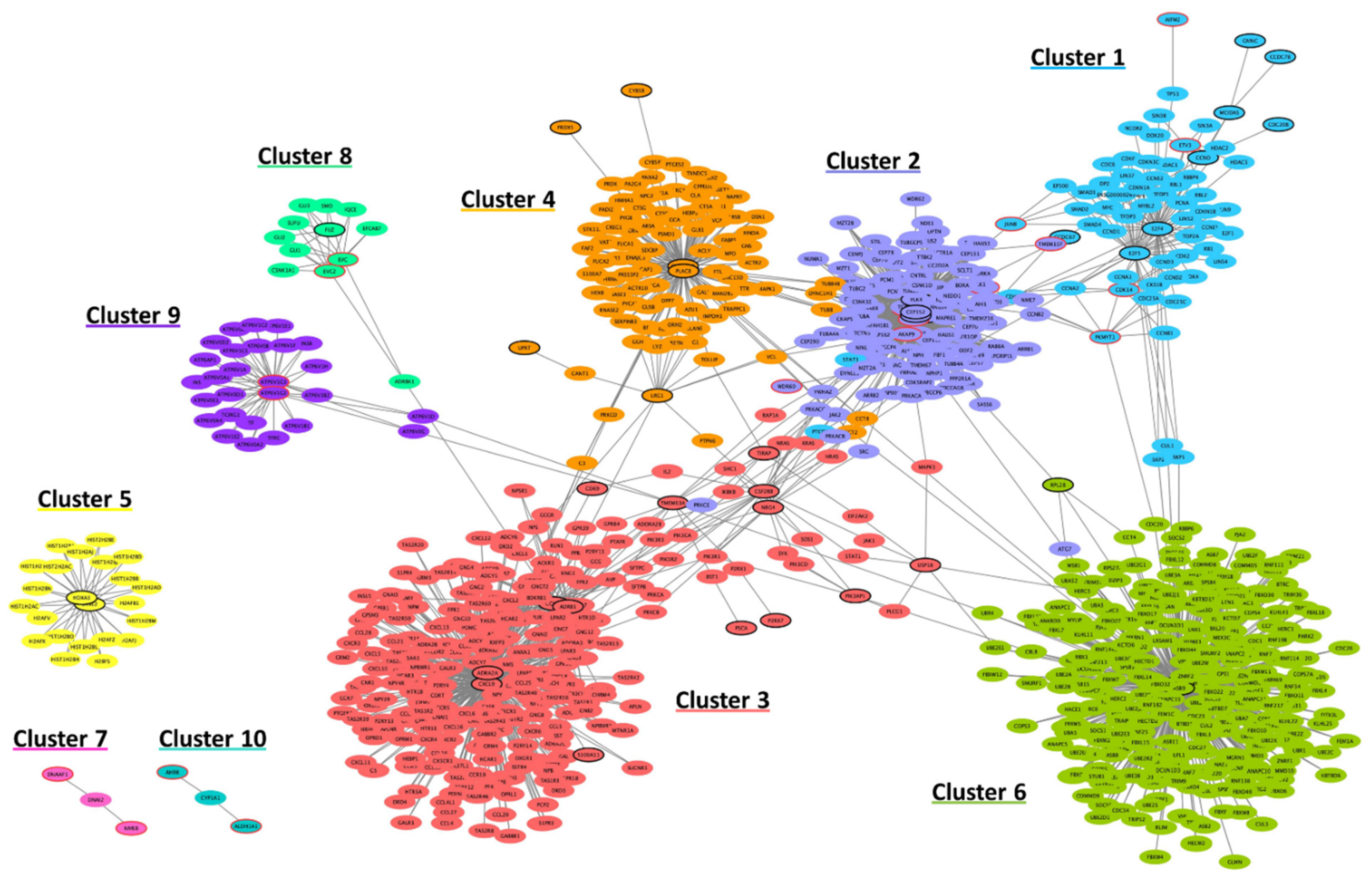
3.3.1. Functional Interaction Networks
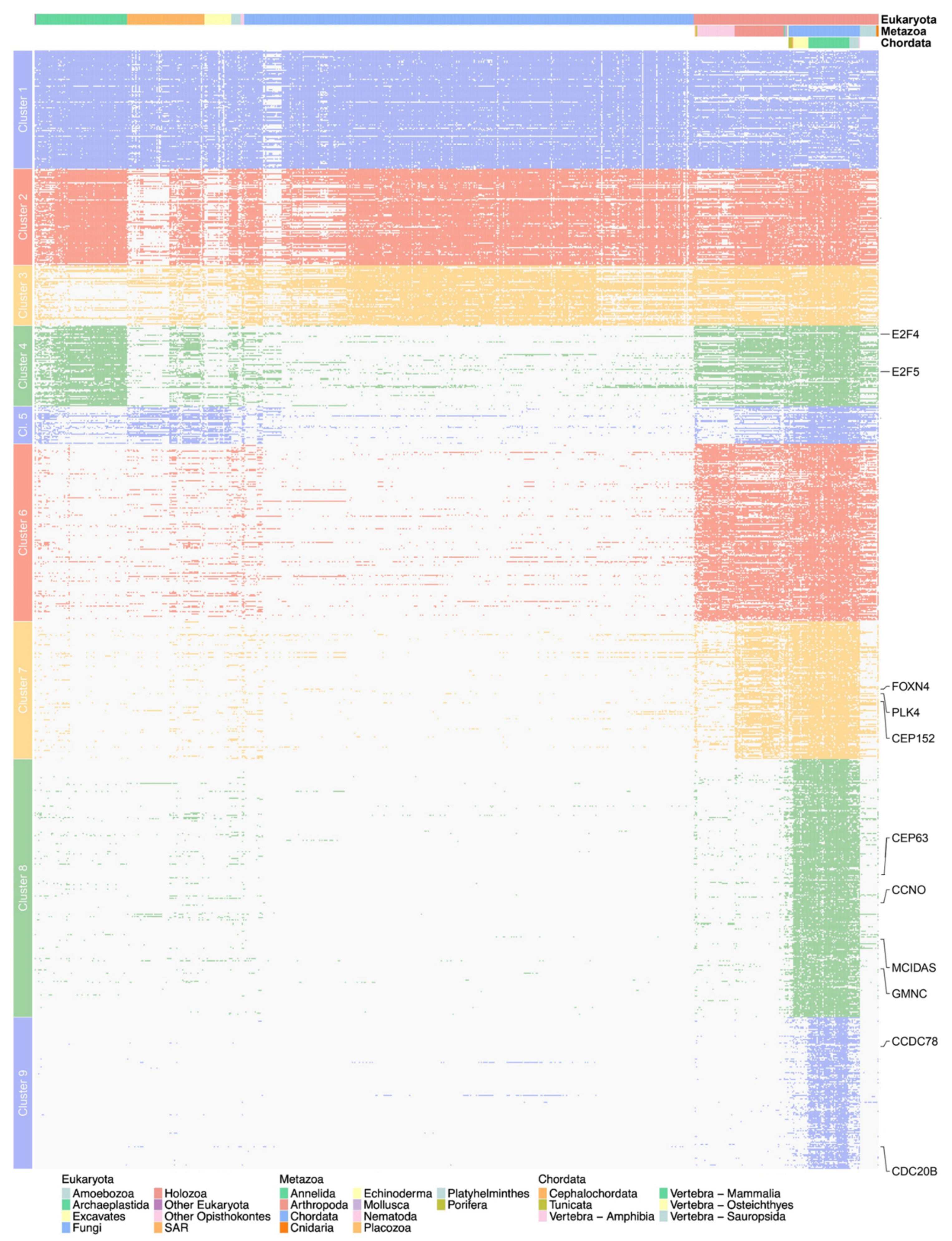
3.3.2. Phylogenetic Profiling of Multiciliated Targets
3.3.3. Identification of Most Promising Candidates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [Green Version]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [Green Version]
- Spassky, N.; Meunier, A. The Development and Functions of Multiciliated Epithelia. Nat. Rev. Mol. Cell Biol. 2017, 18, 423–436. [Google Scholar] [CrossRef]
- Brooks, E.R.; Wallingford, J.B. Multiciliated Cells. Curr. Biol. 2014, 24, R973–R982. [Google Scholar] [CrossRef] [Green Version]
- Kramer-Zucker, A.G.; Olale, F.; Haycraft, C.J.; Yoder, B.K.; Schier, A.F.; Drummond, I.A. Cilia-Driven Fluid Flow in the Zebrafish Pronephros, Brain and Kupffer’s Vesicle Is Required for Normal Organogenesis. Development 2005, 132, 1907–1921. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, C. Animal Evolution: Interrelationships of the Living Phyla, 3rd ed.; Oxford University Press: Oxford, NY, USA, 2012; ISBN 978-0-19-960602-3. [Google Scholar]
- Nikolaev, S.I. Phylogenetic Position of Multicilia Marina and the Evolution of Amoebozoa. Int. J. Syst. Evol. Microbiol. 2006, 56, 1449–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.D. The Morphogenesis of Basal Bodies and Accessory Structures of the Cortex of the Ciliated Protozoan Tetrahymena Pyriformis. J. Cell Biol. 1969, 40, 716–733. [Google Scholar] [CrossRef]
- Machemer, H. Ciliary Activity and the Origin of Metachrony in Paramecium: Effects of Increased Viscosity. J. Exp. Biol. 1972, 57, 239–259. [Google Scholar] [CrossRef]
- Mizukami, I.; Gall, J. Centriole Replication. II. Sperm Formation in the Fern, Marsilea, and the Cycad, Zamia. J. Cell Biol. 1966, 29, 97–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, M.E.; Wickstead, B.; Gull, K.; Langdale, J.A. The Evolution of Land Plant Cilia. New Phytol. 2012, 195, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G. New Animal Phylogeny: Future Challenges for Animal Phylogeny in the Age of Phylogenomics. Org. Divers. Evol. 2016, 16, 419–426. [Google Scholar] [CrossRef]
- Lewis, M.; Stracker, T.H. Transcriptional Regulation of Multiciliated Cell Differentiation. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2021; pp. 51–60. [Google Scholar] [CrossRef]
- Deblandre, G.A.; Wettstein, D.A.; Koyano-Nakagawa, N.; Kintner, C. A Two-Step Mechanism Generates the Spacing Pattern of the Ciliated Cells in the Skin of Xenopus Embryos. Development 1999, 126, 4715–4728. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pathak, N.; Kramer-Zucker, A.; Drummond, I.A. Notch Signaling Controls the Differentiation of Transporting Epithelia and Multiciliated Cells in the Zebrafish Pronephros. Development 2007, 134, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, P.-N.; Vasconcelos, M.; Izvolsky, K.I.; Qian, J.; Lu, J.; Cardoso, W.V. Notch Signaling Controls the Balance of Ciliated and Secretory Cell Fates in Developing Airways. Development 2009, 136, 2297–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcet, B.; Chevalier, B.; Luxardi, G.; Coraux, C.; Zaragosi, L.-E.; Cibois, M.; Robbe-Sermesant, K.; Jolly, T.; Cardinaud, B.; Moreilhon, C.; et al. Control of Vertebrate Multiciliogenesis by MiR-449 through Direct Repression of the Delta/Notch Pathway. Nat. Cell Biol. 2011, 13, 694–701. [Google Scholar] [CrossRef]
- Chu, Q.; Yao, C.; Qi, X.; Stripp, B.R.; Tang, N. STK11 Is Required for the Normal Program of Ciliated Cell Differentiation in Airways. Cell Discov. 2019, 5, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, J.L.; Vladar, E.K.; Axelrod, J.D.; Kintner, C. Multicilin Promotes Centriole Assembly and Ciliogenesis during Multiciliate Cell Differentiation. Nat. Cell Biol. 2012, 14, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Narasimhan, V.; Shboul, M.; Chong, Y.L.; Reversade, B.; Roy, S. Gmnc Is a Master Regulator of the Multiciliated Cell Differentiation Program. Curr. Biol. 2015, 25, 3267–3273. [Google Scholar] [CrossRef] [Green Version]
- Kyrousi, C.; Arbi, M.; Pilz, G.-A.; Pefani, D.-E.; Lalioti, M.-E.; Ninkovic, J.; Go tz, M.; Lygerou, Z.; Taraviras, S. Mcidas and GemC1 Are Key Regulators for the Generation of Multiciliated Ependymal Cells in the Adult Neurogenic Niche. Development 2015, 142, 3661–3674. [Google Scholar] [CrossRef] [Green Version]
- Terré, B.; Piergiovanni, G.; Segura-Bayona, S.; Gil-Gómez, G.; Youssef, S.A.; Attolini, C.S.-O.; Wilsch-Bräuninger, M.; Jung, C.; Rojas, A.M.; Marjanović, M.; et al. GEMC1 Is a Critical Regulator of Multiciliated Cell Differentiation. EMBO J. 2016, 35, 942–960. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Quigley, I.; Omran, H.; Kintner, C. Multicilin Drives Centriole Biogenesis via E2f Proteins. Genes Dev. 2014, 28, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. P73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Nemajerova, A.; Kramer, D.; Siller, S.S.; Herr, C.; Shomroni, O.; Pena, T.; Gallinas Suazo, C.; Glaser, K.; Wildung, M.; Steffen, H.; et al. TAp73 Is a Central Transcriptional Regulator of Airway Multiciliogenesis. Genes Dev. 2016, 30, 1300–1312. [Google Scholar] [CrossRef]
- Tan, F.E.; Vladar, E.K.; Ma, L.; Fuentealba, L.C.; Hoh, R.; Espinoza, F.H.; Axelrod, J.D.; Alvarez-Buylla, A.; Stearns, T.; Kintner, C.; et al. Myb Promotes Centriole Amplification and Later Steps of the Multiciliogenesis Program. Development 2013, 140, 4277–4286. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhu, L.; Zhu, Y.; Cao, J.; Li, S.; Huang, Q.; Xu, T.; Huang, X.; Yan, X.; Zhu, X. The Cep63 Paralogue Deup1 Enables Massive de Novo Centriole Biogenesis for Vertebrate Multiciliogenesis. Nat. Cell Biol. 2013, 15, 1434–1444. [Google Scholar] [CrossRef]
- Mercey, O.; Levine, M.S.; LoMastro, G.M.; Rostaing, P.; Brotslaw, E.; Gomez, V.; Kumar, A.; Spassky, N.; Mitchell, B.J.; Meunier, A.; et al. Massive Centriole Production Can Occur in the Absence of Deuterosomes in Multiciliated Cells. Nat. Cell Biol. 2019, 21, 1544–1552. [Google Scholar] [CrossRef]
- Nanjundappa, R.; Kong, D.; Shim, K.; Stearns, T.; Brody, S.L.; Loncarek, J.; Mahjoub, M.R. Regulation of Cilia Abundance in Multiciliated Cells. Elife 2019, 8, e44039. [Google Scholar] [CrossRef] [PubMed]
- Klos Dehring, D.A.; Vladar, E.K.; Werner, M.E.; Mitchell, J.W.; Hwang, P.; Mitchell, B.J. Deuterosome-Mediated Centriole Biogenesis. Dev. Cell 2013, 27, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, M.C.; Bera, A.N.; Menchen, T.; Kuales, G.; Thriene, K.; Lienkamp, S.S.; Dengjel, J.; Omran, H.; Frank, M.; Arnold, S.J. Cyclin O (Ccno) Functions during Deuterosome-Mediated Centriole Amplification of Multiciliated Cells. EMBO J. 2015, 34, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Huang, T.; Richer, E.J.; Schmidt, J.-E.H.; Zabner, J.; Borok, Z.; Brody, S.L. Role of F-Box Factor Foxj1 in Differentiation of Ciliated Airway Epithelial Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L650–L657. [Google Scholar] [CrossRef] [Green Version]
- Berlucchi, M.; de Santi, M.M.; Bertoni, E.; Spinelli, E.; Timpano, S.; Padoan, R. Ciliary Aplasia Associated with Hydrocephalus: An Extremely Rare Occurrence. Eur. Arch. Oto-Rhino-Laryngol. 2012, 269, 2295–2299. [Google Scholar] [CrossRef] [PubMed]
- Boon, M.; Wallmeier, J.; Ma, L.; Loges, N.T.; Jaspers, M.; Olbrich, H.; Dougherty, G.W.; Raidt, J.; Werner, C.; Amirav, I.; et al. MCIDAS Mutations Result in a Mucociliary Clearance Disorder with Reduced Generation of Multiple Motile Cilia. Nat. Commun. 2014, 5, 4418. [Google Scholar] [CrossRef] [PubMed]
- Wallmeier, J.; Al-Mutairi, D.A.; Chen, C.-T.; Loges, N.T.; Pennekamp, P.; Menchen, T.; Ma, L.; Shamseldin, H.E.; Olbrich, H.; Dougherty, G.W.; et al. Mutations in CCNO Result in Congenital Mucociliary Clearance Disorder with Reduced Generation of Multiple Motile Cilia. Nat. Genet. 2014, 46, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Wallmeier, J.; Frank, D.; Shoemark, A.; Nöthe-Menchen, T.; Cindric, S.; Olbrich, H.; Loges, N.T.; Aprea, I.; Dougherty, G.W.; Pennekamp, P.; et al. De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry. Am. J. Hum. Genet. 2019, 105, 1030–1039. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.P.; Quigley, I.K.; Kintner, C. Foxn4 Promotes Gene Expression Required for the Formation of Multiple Motile Cilia. Development 2016, 143, 4654–4664. [Google Scholar] [CrossRef] [Green Version]
- Quigley, I.K.; Kintner, C. Rfx2 Stabilizes Foxj1 Binding at Chromatin Loops to Enable Multiciliated Cell Gene Expression. PLoS Genet. 2017, 13, e1006538. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Adair-Kirk, T.L.; Patel, A.C.; Huang, T.; Yozamp, N.S.; Xu, J.; Reddy, E.P.; Byers, D.E.; Pierce, R.A.; Holtzman, M.J.; et al. Myb Permits Multilineage Airway Epithelial Cell Differentiation. Stem Cells 2014, 32, 3245–3256. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Hazan, R.; Danielian, P.S.; Mahoney, J.E.; Li, H.; Lu, J.; Miller, E.S.; Zhu, X.; Lees, J.A.; Cardoso, W.V. Cytoplasmic E2f4 Forms Organizing Centres for Initiation of Centriole Amplification during Multiciliogenesis. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Karimi, K.; Fortriede, J.D.; Lotay, V.S.; Burns, K.A.; Wang, D.Z.; Fisher, M.E.; Pells, T.J.; James-Zorn, C.; Wang, Y.; Ponferrada, V.G.; et al. Xenbase: A Genomic, Epigenomic and Transcriptomic Model Organism Database. Nucleic Acids Res. 2018, 46, D861–D868. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plewniak, F.; Bianchetti, L.; Brelivet, Y.; Carles, A.; Chalmel, F.; Lecompte, O.; Mochel, T.; Moulinier, L.; Muller, A.; Muller, J.; et al. PipeAlign: A New Toolkit for Protein Family Analysis. Nucleic Acids Res. 2003, 31, 3829–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.H.; Kuznetsov, A.; Ananiev, V.; Asztalos, A.; Borodin, E.; Evgeniev, V.; Joukov, V.; Lotov, V.; Pannu, R.; Rudnev, D.; et al. Accessing NCBI Data Using the NCBI Sequence Viewer and Genome Data Viewer (GDV). Genome Res. 2021, 31, 159–169. [Google Scholar] [CrossRef]
- Defosset, A.; Kress, A.; Nevers, Y.; Ripp, R.; Thompson, J.D.; Poch, O.; Lecompte, O. Proteome-Scale Detection of Differential Conservation Patterns at Protein and Subprotein Levels with BLUR. Genome Biol. Evol. 2020, 13, evaa248. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community Structure Analysis of Biological Networks. Bioinformatics 2010, 26, 3135–3137. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. ClusterMaker: A Multi-Algorithm Clustering Plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [Green Version]
- Nevers, Y.; Kress, A.; Defosset, A.; Ripp, R.; Linard, B.; Thompson, J.D.; Poch, O.; Lecompte, O. OrthoInspector 3.0: Open Portal for Comparative Genomics. Nucleic Acids Res. 2019, 47, D411–D418. [Google Scholar] [CrossRef]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER Version 14: More Genomes, a New PANTHER GO-Slim and Improvements in Enrichment Analysis Tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Huang, K. Transport of Ciliary Membrane Proteins. Front. Cell Dev. Biol. 2020, 7, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Rayamajhi, D.; Ravi, V.; Narasimhan, V.; Chong, Y.L.; Lu, H.; Venkatesh, B.; Roy, S. Conservation as Well as Divergence in Mcidas Function Underlies the Differentiation of Multiciliated Cells in Vertebrates. Dev. Biol. 2020, 465, 168–177. [Google Scholar] [CrossRef]
- Honda, S.; Shimizu, S. Autophagy Controls Centrosome Number. Oncotarget 2017, 8, 14277–14278. [Google Scholar] [CrossRef] [Green Version]
- Marra, A.N.; Cheng, C.N.; Adeeb, B.; Addiego, A.; Wesselman, H.M.; Chambers, B.E.; Chambers, J.M.; Wingert, R.A. Iroquois Transcription Factor Irx2a Is Required for Multiciliated and Transporter Cell Fate Decisions during Zebrafish Pronephros Development. Sci. Rep. 2019, 9, 6454. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Hong, K.-M.; Yang, S.-H.; Chowdhuri, S.R.; Player, A.; Hames, M.; Fukuoka, J.; Meerzaman, D.; Dracheva, T.; Sun, Z.; Yang, P.; et al. Inactivation of LLC1 Gene in Nonsmall Cell Lung Cancer. Int. J. Cancer 2007, 120, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Qiao, Y.; Di, Q.; Le, X.; Zhang, L.; Zhang, X.; Zhang, C.; Cheng, J.; Zong, S.; Koide, S.S.; et al. Interaction of SH3P13 and DYDC1 Protein: A Germ Cell Component That Regulates Acrosome Biogenesis during Spermiogenesis. Eur. J. Cell Biol. 2009, 88, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, Z.; Rodriguez, S.M.B.; Vardarajan, B.N.; Renton, A.E.; Goate, A.M.; Mayeux, R.; Wang, G.T.; Leal, S.M. A Quantitative Trait Rare Variant Nonparametric Linkage Method with Application to Age-at-Onset of Alzheimer’s Disease. Eur. J. Hum. Genet. 2020, 28, 1734–1742. [Google Scholar] [CrossRef]
- Raybould, R.; Sims, R. Searching the Dark Genome for Alzheimer’s Disease Risk Variants. Brain Sci. 2021, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Zeiske, E. Development of the Olfactory Organ in the Zebrafish, Brachydanio Rerio. J. Comp. Neurol. 1993, 333, 289–300. [Google Scholar] [CrossRef]
- Nevers, Y.; Prasad, M.K.; Poidevin, L.; Chennen, K.; Allot, A.; Kress, A.; Ripp, R.; Thompson, J.D.; Dollfus, H.; Poch, O.; et al. Insights into Ciliary Genes and Evolution from Multi-Level Phylogenetic Profiling. Mol. Biol. Evol. 2017, 34, 2016–2034. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Overall Experimental Design (Multiciliation vs. No Multiciliation) | Species | Experiment Type |
|---|---|---|---|
| GSE32452 [19] | Notch intracellular domain (ICD) + glucocorticoid inducible Multicilin vs. ICD | Xenopus laevis | Microarray |
| GSE59309 [23] | Inducible Multicilin vs. inducible Multicilin + truncated E2F4 | X. laevis | RNASeq |
| GSE89271 [38] | Inducible Multicilin vs. inducible Multicilin + Foxn4 morpholino | X. laevis | RNASeq |
| Inducible Multicilin vs. inducible Multicilin + CRISPR/Cas9 Foxn4 mutant | X. laevis | RNASeq | |
| Inducible Multicilin vs. inducible Multicilin + CRISPR/Cas9 Foxj1 mutant | X. laevis | RNASeq | |
| GSE76342 [39] | Notch- vs. ICD; ICD vs. ICD + Multicilin; Notch- vs. Notch- + Multicilin- | X. laevis | RNASeq |
| GSE60365 [40] | Non-targeted shRNA vs. Myb shRNA | Mus musculus | Microarray |
| GSE75715 [25] | Wild Type vs. p73 knockout | M. musculus | RNASeq |
| GSE73331 [41] | Wild Type vs. E2F4 knockout | M. musculus | Microarray |
| GSE116690 [18] | Stk11+ vs. Stk11- | M. musculus | RNASeq |
| Clusters | Characteristics | Genes | GO Terms | p-Value |
|---|---|---|---|---|
| 3, 6, 7, 14 | Foxj1/Foxn4 targets | 759 | ‘establishment of localization’ | 1.64 × 10−13 |
| 17, 18, 19, 26, 27 | MCIDAS/E2F4 complex and Foxj1 or Foxn4 targets | 308 | ‘cilium organization’ | 8.28 × 10−23 |
| 22, 24, 25, 28 | Mouse genes | 160 | ‘cilium movement’ | 1.32 × 10−9 |
| 20 | MCIDAS/E2F4 complex targets | 58 | ‘centrosome cycle’ | 1.30 × 10−13 |
| 8, 9, 12, 13 | Multiciliated clusters | 587 | ‘cilium organization’ | 1.94 × 10−92 |
| Gene | Transcriptomics Cluster | BLUR | STRING Cluster | Profiling Cluster |
|---|---|---|---|---|
| C1orf189 | 13 | Absent Otomorpha | - | 9 |
| C20orf85 | 18 | Absent Otomorpha | - | 9 |
| C5orf24 | 3 | Mildly likely divergence | - | 8 |
| KIAA1841 | 19 | Mildly likely divergence | - | 5 |
| FAM181A | 9 | Highly likely divergence | - | 8 |
| IQCK | 12 | Mildly likely divergence | - | 7 |
| LRRC43 | 22 | Mildly likely divergence | - | 8 |
| DYDC1 | 8 | Mildly likely divergence | - | 8 |
| CFAP47 | 12 | Absent Otomorpha | - | 5 |
| ANKRD60 | 17 | Absent Otomorpha | - | 9 |
| TEX43 | 18 | Absent Otomorpha | - | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Defosset, A.; Merlat, D.; Poidevin, L.; Nevers, Y.; Kress, A.; Poch, O.; Lecompte, O. Novel Approach Combining Transcriptional and Evolutionary Signatures to Identify New Multiciliation Genes. Genes 2021, 12, 1452. https://doi.org/10.3390/genes12091452
Defosset A, Merlat D, Poidevin L, Nevers Y, Kress A, Poch O, Lecompte O. Novel Approach Combining Transcriptional and Evolutionary Signatures to Identify New Multiciliation Genes. Genes. 2021; 12(9):1452. https://doi.org/10.3390/genes12091452
Chicago/Turabian StyleDefosset, Audrey, Dorine Merlat, Laetitia Poidevin, Yannis Nevers, Arnaud Kress, Olivier Poch, and Odile Lecompte. 2021. "Novel Approach Combining Transcriptional and Evolutionary Signatures to Identify New Multiciliation Genes" Genes 12, no. 9: 1452. https://doi.org/10.3390/genes12091452
APA StyleDefosset, A., Merlat, D., Poidevin, L., Nevers, Y., Kress, A., Poch, O., & Lecompte, O. (2021). Novel Approach Combining Transcriptional and Evolutionary Signatures to Identify New Multiciliation Genes. Genes, 12(9), 1452. https://doi.org/10.3390/genes12091452