
Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skeletal Samples
2.2. DNA Extraction
2.3. Library Preparation
2.3.1. In House MPI Method
2.3.2. SRSLY Kit
2.3.3. KAPA Hyper Prep Kit
2.3.4. Quantification
2.4. MtDNA Hybridization Capture
2.5. Sequencing and Data Processing
3. Results
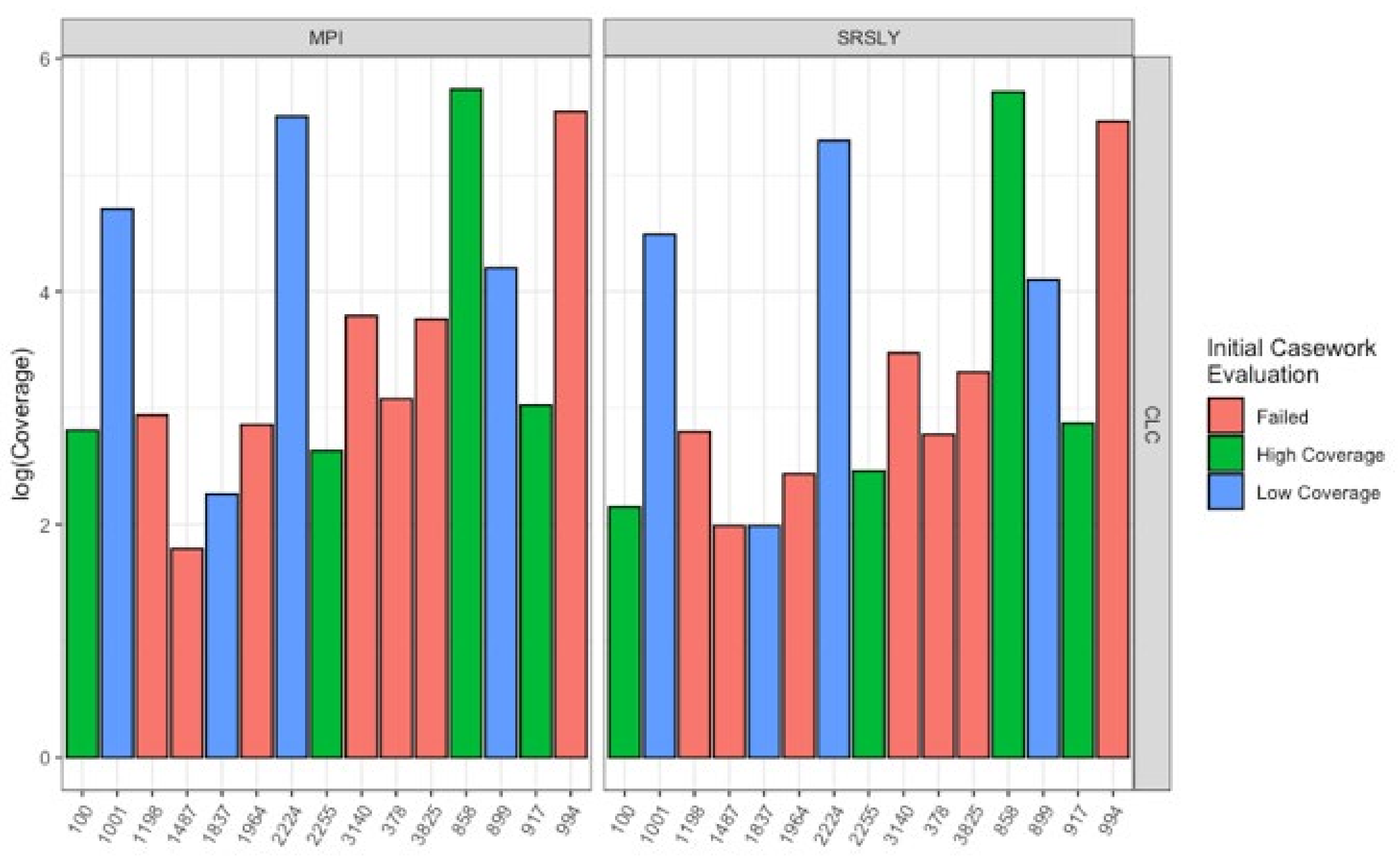
3.1. Evaluation of Extraction Methods
3.2. Characteristics of Recovered DNA
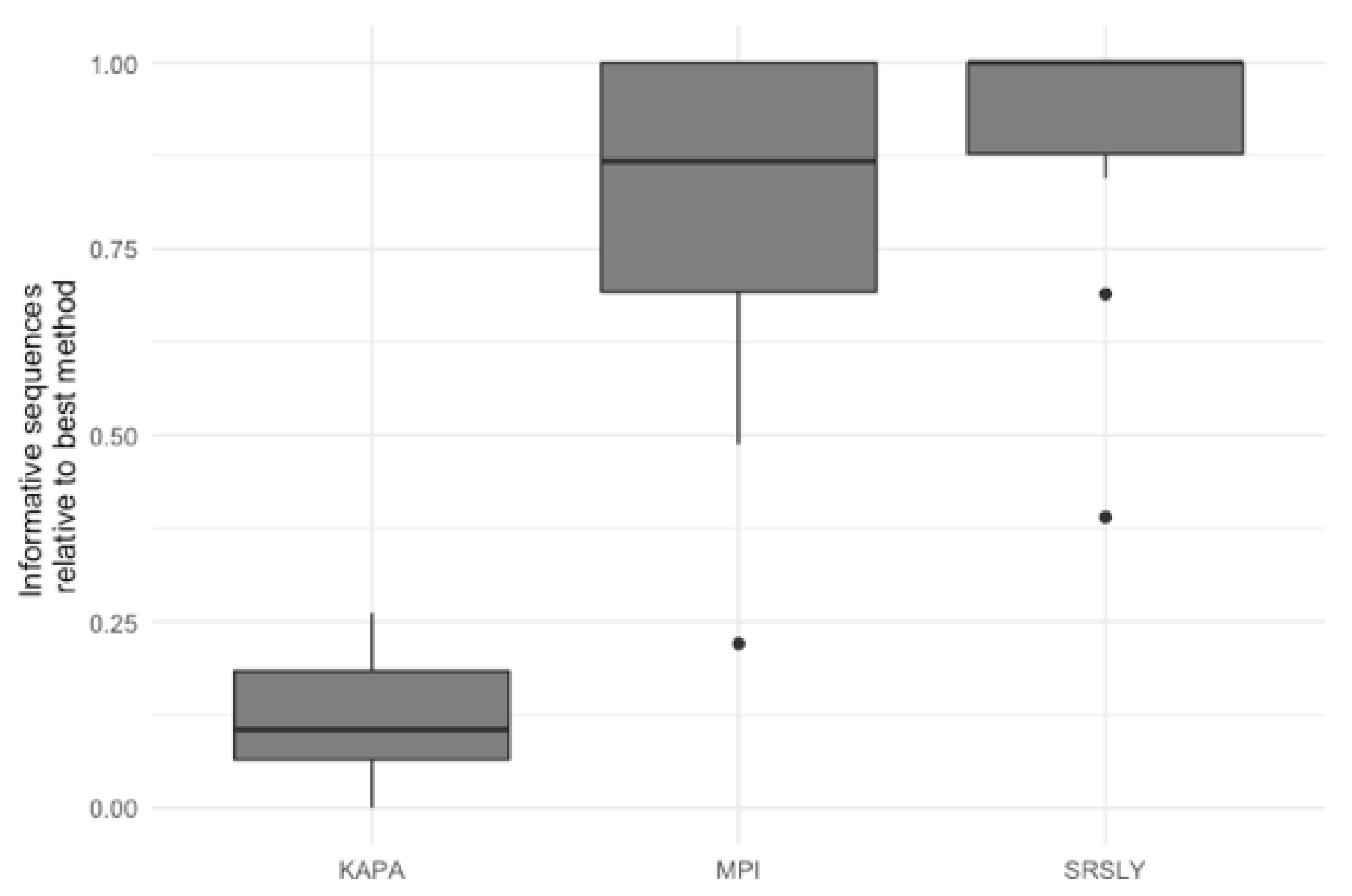
3.3. Evaluation of Library Preparartion Protocols
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- First Criminal Conviction Secured with Next-Gen Forensic DNA Technology. 2019. Available online: https://verogen.com/first-criminal-conviction-with-next-gen-forensic-dna/ (accessed on 2 December 2021).
- National DNA Index System (NDIS) Operational Procedures Manual; Version 10; FBI Laboratory: Quantico, VA, USA, 2021.
- Greytak, E.M.; Moore, C.; Armentrout, S.L. Genetic genealogy for cold case and active investigations. Forensic Sci. Int. 2019, 299, 103–113. [Google Scholar] [CrossRef]
- Marshall, C.; Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, A.; Fernandes, T.; Taveira, N. Mitochondrial DNA in human identification: A review. PeerJ 2019, 7, e7314. [Google Scholar] [CrossRef]
- Ambers, A.; Bus, M.M.; King, J.L.; Jones, B.; Durst, J.; Bruseth, J.E.; Gill-King, H.; Budowle, B. Forensic genetic investigation of human skeletal remains recovered from the La Belle shipwreck. Forensic Sci. Int. 2020, 306, 110050. [Google Scholar] [CrossRef]
- Cole, P.M. POW/MIA Issues: The Korean War; NDRI: Santa Monica, CA, USA, 1994; Volume 1. [Google Scholar]
- Keene, J. Bodily matters above and below ground: The treatment of American remains from the Korean war. Public Hist. 2010, 32, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Morris-Suzuki, T. Lavish are the dead: Re-envisioning Japan’s Korean war. Asia Pac. J. Jpn. Focus 2013, 11, 1–8. [Google Scholar]
- Coleman, B.L. Recovering the Korean war dead, 1950–1958: Graves registration, forensic anthropology, and wartime memorialization. J. Mil. Hist. 2008, 72, 179–222. [Google Scholar] [CrossRef]
- Gilbert, M.T.P.; Haselkorn, T.; Bunce, M.; Sanchez, J.J.; Lucas, S.B.; Jewell, L.D.; Van Marck, E.; Worobey, M. The isolation of nucleic acids from fixed, paraffin-embedded tissues-which methods are useful when? PLoS ONE 2007, 2, e537. [Google Scholar] [CrossRef]
- Kistler, L.; Ware, R.; Smith, O.; Collins, M.; Allaby, R.G. A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. 2017, 45, 6310–6320. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.; Byrd, J.; Sava, V. Joint POW/MIA Accounting Command’s Central Identification. Forensic Anthropol. Lab. 2008, 47, 63–80. [Google Scholar]
- Edson, S.; Ross, J.P.; Coble, M.D.; Parsons, T.J.; Barritt, S.M. Naming the dead—Confronting the realities of rapid identification of degraded skeletal remains. Forensic Sci. Rev. 2004, 16, 63–90. [Google Scholar] [PubMed]
- Gorden, E.M.; Greytak, E.M.; Sturk-Andreaggi, K.; Cady, J.; McMahon, T.P.; Armentrout, S.; Marshall, C. Extended kinship analysis of historical remains using SNP capture. Forensic Sci. Int. Genet. 2022, 57, 102636. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [Green Version]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef]
- Gansauge, M.-T.; Aximu-Petri, A.; Nagel, S.; Meyer, M. Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA. Nat. Protoc. 2020, 15, 2279–2300. [Google Scholar] [CrossRef]
- Fu, Q.; Meyer, M.; Gao, X.; Stenzel, U.; Burbano, H.A.; Kelso, J.; Pääbo, S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl. Acad. Sci. USA 2013, 110, 2223–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchan, T.; Kusliy, M.A.; Khan, N.; Chauvey, L.; Tonasso-Calvière, L.; Schiavinato, S.; Southon, J.; Keller, M.; Kitagawa, K.; Krause, J.; et al. Performance and automation of ancient DNA capture with RNA hyRAD probes. Mol. Ecol. Resour. 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Zavala, E.I.; Gómez-Olivencia, A.; Jacobs, Z.; Slon, V.; Mafessoni, F.; Romagné, F.; Pearson, A.; Petr, M.; Sala, N.; et al. Unearthing Neanderthal population history using nuclear and mitochondrial DNA from cave sediments. Science 2021, 372, eabf1667. [Google Scholar] [CrossRef] [PubMed]
- Zavala, E.I.; Jacobs, Z.; Vernot, B.; Shunkov, M.V.; Kozlikin, M.B.; Derevianko, A.P.; Essel, E.; de Fillipo, C.; Nagel, S.; Richter, J.; et al. Pleistocene sediment DNA reveals hominin and faunal turnovers at Denisova Cave. Nature 2021, 595, 399–403. [Google Scholar] [CrossRef]
- Meyer, M.; Arsuaga, J.L.; de Filippo, C.; Nagel, S.; Aximu-Petri, A.; Nickel, B.; Martinez, I.; Gracia, A.; de Castro, J.M.B.; Carbonell, E.; et al. Nuclear DNA sequences from the middle pleistocene sima de los huesos hominins. Nature 2016, 531, 504–507. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M.; Gansauge, M.-T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prüfer, K.; De Filippo, C.; et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Stiller, M.; Sucker, A.; Griewank, K.; Aust, D.; Baretton, G.B.; Schadendorf, D.; Horn, S. Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) cancer DNA. Oncotarget 2016, 7, 59115–59128. [Google Scholar] [CrossRef] [Green Version]
- Gansauge, M.-T.; Gerber, T.; Glocke, I.; Korlević, P.; Lippik, L.; Nagel, S.; Riehl, L.M.; Schmidt, A.; Meyer, M. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 2017, 45, e79. [Google Scholar]
- Straube, N.; Lyra, M.L.; Paijmans, J.L.A.; Preick, M.; Basler, N.; Penner, J.; Rödel, M.; Westbury, M.V.; Haddad, C.F.B.; Barlow, A.; et al. Successful application of ancient DNA extraction and library construction protocols to museum wet collection specimens. Mol. Ecol. Resour. 2021, 21, 2299–2315. [Google Scholar] [CrossRef]
- Zavala, E.I.; Rajagopal, S.; Perry, G.H.; Kruzic, I.; Bašić, Ž.; Parsons, T.J.; Holland, M.M. Impact of DNA degradation on massively parallel sequencing-based autosomal STR, iiSNP, and mitochondrial DNA typing systems. Int. J. Legal. Med. 2019, 133, 1369–1380. [Google Scholar] [CrossRef]
- Hofreiter, M.; Sneberger, J.; Pospisek, M.; Vanek, D. Progress in forensic bone DNA analysis: Lessons learned from ancient DNA. Forensic Sci. Int. Genet. 2021, 54, 102538. [Google Scholar] [CrossRef]
- Xavier, C.; Eduardoff, M.; Bertoglio, B.; Amory, C.; Berger, C.; Casas-Vargas, A.; Pallua, J.; Parson, W. Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age. Genes 2021, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Edson, S.M. Extraction of DNA from Skeletonized Postcranial Remains: A Discussion of Protocols and Testing Modalities. J. Forensic Sci. 2019, 64, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Gorden, E.M.; Sturk-Andreaggi, K.; Marshall, C. Repair of DNA damage caused by cytosine deamination in mitochondrial DNA of forensic case samples. Forensic Sci. Int. Genet. 2018, 34, 257–264. [Google Scholar] [CrossRef] [Green Version]
- SRSLY NGS Library Prep Kit for Illumina PicoPlus and NanoPlus UDIs and UMIs/UDIs: Unabridged Instruction Manual; Clarert Biosience, LLC.: Santa Cruz, CA, USA, 2020.
- Renaud, G.; Stenzel, U.; Kelso, J. leeHom: Adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res. 2014, 42, e141. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Peck, M.A.; Boysen, C.; Dekker, P.; McMahon, T.P.; Marshall, C.K. AQME: A forensic mitochondrial DNA analysis tool for next-generation sequencing data. Forensic Sci. Int. Genet. 2017, 31, 189–197. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Parson, W.; Allen, M.; Marshall, C. Impact of the sequencing method on the detection and interpretation of mitochondrial DNA length heteroplasmy. Forensic Sci. Int. Genet. 2020, 44, 102205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slon, V.; Hopfe, C.; Weiß, C.L.; Mafessoni, F.; de la Rasilla, M.; Lalueza-Fox, C.; Rosas, A.; Soressi, M.; Knul, M.V.; Miller, R.; et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 2017, 356, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.; Kelso, J.; Prufer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyregne, S.; Peter, B.M. AuthentiCT: A model of ancient DNA damage to estimate the proportion of present-day DNA contamination. Genome Biol. 2020, 21, 246. [Google Scholar] [CrossRef]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Pääbo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar]
- Rathbun, M.M.; McElhoe, J.A.; Parson, W.; Holland, M.M. Considering DNA damage when interpreting mtDNA heteroplasmy in deep sequencing data. Forensic Sci. Int. Genet. 2017, 26, 1–11. [Google Scholar] [CrossRef]
- Holland, C.A.; McElhoe, J.A.; Gaston-Sanchez, S.; Holland, M.M. Damage patterns observed in mtDNA control region MPS data for a range of template concentrations and when using different amplification approaches. Int. J. Leg. Med. 2021, 135, 91–106. [Google Scholar] [CrossRef]
- Hajdinjak, M.; Mafessoni, F.; Skov, L.; Vernot, B.; Hübner, A.; Fu, Q.; Essel, E.; Nagel, S.; Nickel, B.; Richter, J.; et al. Initial Upper Palaeolithic humans in Europe had recent Neanderthal ancestry. Nature 2021, 592, 253–257. [Google Scholar] [CrossRef]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.; Taylor, R.; Sturk-Andreaggi, K.; Barritt-Ross, S.; Berg, G.E.; McMahon, T.P. Mitochondrial DNA haplogrouping to assist with the identification of unknown service members from the World War II Battle of Tarawa. Forensic Sci. Int. Genet. 2020, 47, 102291. [Google Scholar] [CrossRef] [PubMed]
- Troll, C.J.; Kapp, J.; Rao, V.; Harkins, K.; Cole, C.; Naughton, C.; Morgan, J.M.; Shapiro, B.; Green, R.E. A ligation-based single-stranded library preparation method to analyze cell-free DNA and synthetic oligos. BMC Genom. 2019, 2, 1023. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.-Y.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Fu, Q.; Aximu-Petri, A.; Glocke, I.; Nickel, B.; Arsuaga, J.L.; Martinez, I.; Gracia, A.; De Castro, J.M.B.; Carbonell, E.; et al. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 2014, 505, 403–406. [Google Scholar] [CrossRef]
- Hajdinjak, M.; Fu, Q.; Hübner, A.; Petr, M.; Mafessoni, F.; Grote, S.; Skoglund, P.; Narasimham, V.; Rougier, H.; Crevecoeur, I.; et al. Reconstructing the genetic history of late Neanderthals. Nature 2018, 555, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Korlević, P.; Gerber, T.; Gansauge, M.-T.; Hajdinjak, M.; Nagel, S.; Aximu-Petri, A.; Meyer, M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 2015, 59, 87–93. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Skeletal Element | Context | Cemetery of Disinterment | Formalin Treatment | Powder Application | Casework Results |
|---|---|---|---|---|---|---|
| 2224 | Tibia | WWII-Tarawa | NMCP 1 | Unknown | Not visible | High Coverage |
| 0858 | Temporal | WWII-USS Oklahoma | NMCP 1 | No, Oil Soaked | Likely | High Coverage |
| 0994 | Femur | WWII-Buna | MAC 2 | Unknown | Not visible | High Coverage |
| 2255 | Humerus | WWII-Cabanatuan | MAC 2 | Unknown | Not visible | High Coverage |
| 0899 | Humerus | WWII-Tarawa | NMCP 1 | Unknown | Yes | Low Coverage |
| 0100 | Femur | Korea | NMCP 1 | Likely | Yes | Low Coverage |
| 1001 | Os coxa | WWII-Yugoslavia | SRAC 3 | Unknown | Not visible | Low Coverage |
| 3140 | Radius | WWII-Burma | NMCP 1 | Unknown | Yes | Low Coverage |
| 0378 | Femur | WWII-Tarawa | NMCP 1 | Unknown | Yes | Failed |
| 1837 | Tibia | WWII-Tarawa | NMCP 1 | Unknown | Yes | Failed |
| 1198 | Tibia | Korea | NMCP 1 | Likely | Yes | Failed |
| 1964 | Humerus | Korea | NMCP 1 | Likely | Yes | Failed |
| 0917 | Tibia | Korea | NMCP 1 | Likely | Yes | Failed |
| 3825 | Radius | Korea | NMCP 1 | Likely | Yes | Failed |
| 1487 | Femur | WWII-Italy | SRAC 3 | Unknown | Yes | Failed |
| Method | Bone Powder (g) | Digestion Buffer | Digestion Buffer Volume (mL) | Proteinase K (20 mg/mL) (µL) | Incubation Temperature (°C) | DNA Purification | Repair Protocol |
|---|---|---|---|---|---|---|---|
| AFDIL (PM) | 1.0 | Demin buffer 1 | 7.5 | 200 | 56 | PCIA with buffer exchange | NA (MinElute purification) |
| AFDIL-USER (PUM) | 1.0 | Demin buffer 1 | 7.5 | 200 | 56 | PCIA with buffer exchange | USER (NEB, Ipswich, MA, USA) |
| AFDIL-FFPE (PFM) | 1.0 | Demin buffer 1 | 7.5 | 200 | 56 | PCIA with buffer exchange | NEBNext FFPE DNA Repair Mix (NEB) |
| Dabney—37 (aDNA37) | 0.2 | Dabney buffer 2 | 1.0 | 25 | 37 | Silica column and PB Buffer | NA |
| Dabney—56 (aDNA56) | Dabney 37 remaining pellet | Dabney buffer 2 | 1.0 | 25 | 56 | Silica column and PB Buffer | NA |
| AFDIL-Dabney (A_D) | 0.2 | Demin buffer 1 | 4.0 | 200 | 56 | Silica column and PB Buffer | NA |
| Sample ID | Casework | MPI | SRSLY (Pig Out) | |||||
|---|---|---|---|---|---|---|---|---|
| Reported Bases | Predicted Haplogroup | Reported Bases | Predicted Haplogroup | Discordant Sites (Compared to Casework) | Reported Bases | Predicted Haplogroup | Discordant Sites (Compared to Casework) | |
| High Coverage | ||||||||
| 2224 | 16507 | H18 | 16569 | H18 | 0 | 16568 | H18 | 0 |
| 0858 | 16507 | V3c | 16568 | V3c | 0 | 16569 | V3c | 0 |
| 0994 | 16507 | J1c4b | 16569 | J1c4b | 0 | 16569 | J1c4b | 0 |
| 2255 | 16507 | J1c3 | 16268 | J1c3/J1c3h/J1c3k | 1 | 16144 | J1c3 | 1 |
| Low Coverage | ||||||||
| 0899 | 16507 | H4a1a4b | 16565 | H4a1a4b | 0 | 16563 | H4a1a4b | 0 |
| 0100 | 16183 | H24a | 16181 | H24a | 0 | 13585 | H24/H24a | 0 |
| 1001 | 15765 | H1c6 | 16566 | H1c6 | 0 | 16567 | H1c6 | 0 |
| 3140 | 16505 | U5b1b1-16192 | 16555 | U5b1b1- 16192 | 0 | 16515 | U5b1b1-16192 | 0 |
| Failed in Casework | ||||||||
| 0378 | NA | NA | 15951 | U5a1a1+16362/U5a1a1d/U5a1a1d1 | NA | 15681 | U5a1a1+16362/U5a1a1d/U5a1a1d1 | NA |
| 1837 | NA | NA | 13504 | K1c2 | NA | 11526 | K1c2 | NA |
| 1198 | NA | NA | 16091 | H1c3b | NA | 15821 | H1c3b | NA |
| 1964 | NA | NA | 16010 | L2a1c+16086/L2a1c3b | NA | 14871 | L2a1c+16086 | NA |
| 0917 | NA | NA | 16386 | K1a1b2b | NA | 16004 | K1a1b2b | NA |
| 3825 | NA | NA | 16550 | H11a | NA | 16319 | H11a | NA |
| 1487 | NA | NA | 10715 | K1a (and subhaplogroups of K1a) | NA | 12009 | K1a9 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavala, E.I.; Thomas, J.T.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Meyers, K.K.; Barrit-Ross, S.; Aximu-Petri, A.; Richter, J.; Nickel, B.; Berg, G.E.; et al. Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns. Genes 2022, 13, 129. https://doi.org/10.3390/genes13010129
Zavala EI, Thomas JT, Sturk-Andreaggi K, Daniels-Higginbotham J, Meyers KK, Barrit-Ross S, Aximu-Petri A, Richter J, Nickel B, Berg GE, et al. Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns. Genes. 2022; 13(1):129. https://doi.org/10.3390/genes13010129
Chicago/Turabian StyleZavala, Elena I., Jacqueline Tyler Thomas, Kimberly Sturk-Andreaggi, Jennifer Daniels-Higginbotham, Kerriann K. Meyers, Suzanne Barrit-Ross, Ayinuer Aximu-Petri, Julia Richter, Birgit Nickel, Gregory E. Berg, and et al. 2022. "Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns" Genes 13, no. 1: 129. https://doi.org/10.3390/genes13010129
APA StyleZavala, E. I., Thomas, J. T., Sturk-Andreaggi, K., Daniels-Higginbotham, J., Meyers, K. K., Barrit-Ross, S., Aximu-Petri, A., Richter, J., Nickel, B., Berg, G. E., McMahon, T. P., Meyer, M., & Marshall, C. (2022). Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns. Genes, 13(1), 129. https://doi.org/10.3390/genes13010129