
A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
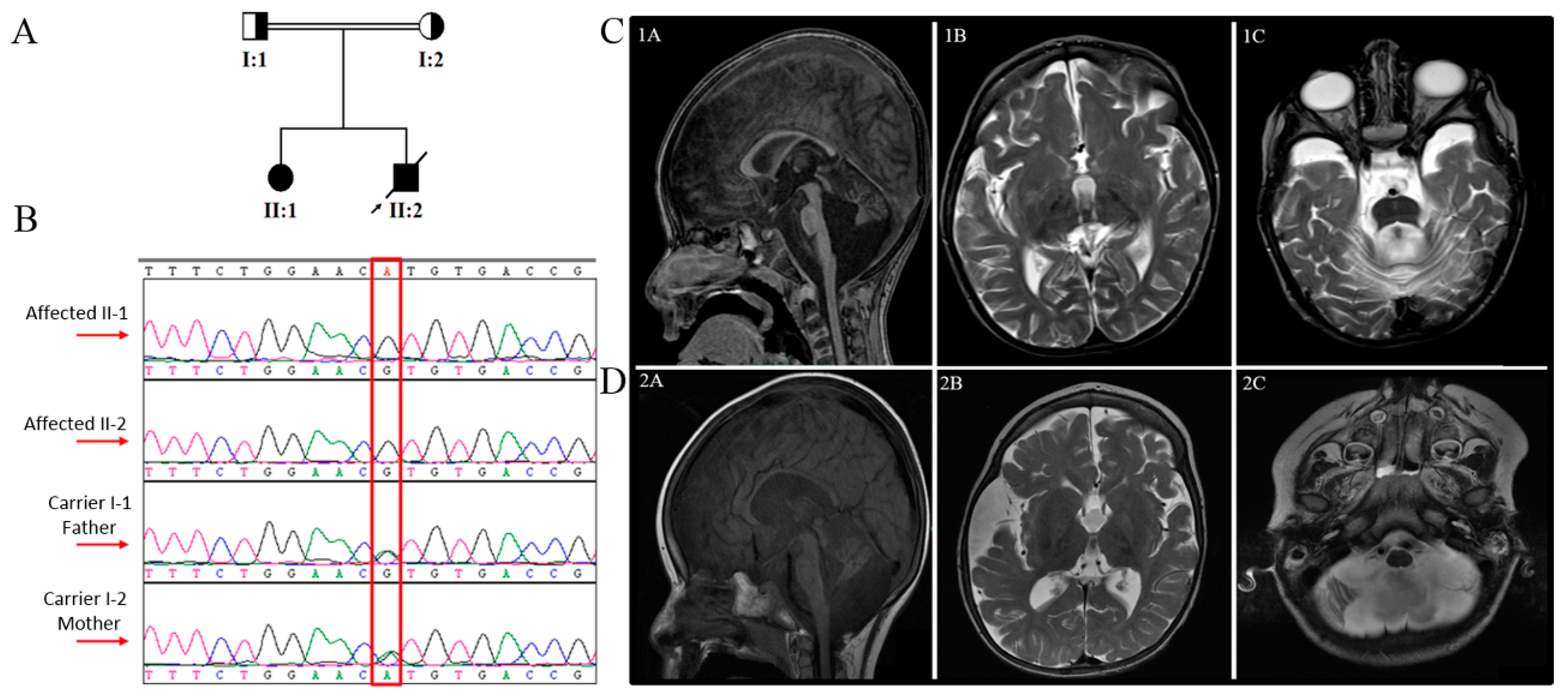
2.1. Patients and Ethics
2.2. DNA Isolation, PCR, Sanger Sequencing
2.3. Whole Exome Sequencing (WES) and Variant Detection
2.4. In Silico Pathogenicity Prediction Analyses
2.5. Computational Structural Modeling and Analysis of the Novel Variant
2.6. Tandem Mass Spectrometry
3. Results
3.1. Clinical Findings
3.1.1. Family A, Patient 1
3.1.2. Family A, Patient 2
3.2. Exome Sequencing and In Silico Functional Prediction Analyses
3.3. Computational Structural Modeling and Analysis of the Novel Variant
4. Discussion
5. List of the Databases
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Yong, J.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin4a novel component of the SMN complex that is found in both gems and nucleoli. J. Cell Biol. 2000, 148, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Lanfranco, M.; Cacciottolo, R.; Borg, R.M.; Vassallo, N.; Juge, F.; Bordonné, R.; Cauchi, R.J. Novel interactors of the Drosophila Survival Motor Neuron (SMN) Complex suggest its full conservation. FEBS Lett. 2017, 591, 3600–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matera, A.G.; Raimer, A.C.; Schmidt, C.A.; Kelly, J.A.; Droby, G.N.; Baillat, D.; Have, S.T.; Lamond, A.I.; Wagner, E.J.; Gray, K.M. Composition of the Survival Motor Neuron (SMN) Complex in Drosophila melanogaster. G3 Genes|Genomes|Genet. 2019, 9, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Meier, I.D.; Walker, M.P.; Matera, A.G. Gemin4 is an essential gene in mice, and its overexpression in human cells causes relocalization of the SMN complex to the nucleoplasm. Biol. Open 2018, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Mouillet, J.-F.; Yan, X.; Ou, Q.; Jin, L.; Muglia, L.J.; Crawford, P.A.; Sadovsky, Y. DEAD-Box Protein-103 (DP103, Ddx20) Is Essential for Early Embryonic Development and Modulates Ovarian Morphology and Function. Endocrinology 2008, 149, 2168–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2018, 21, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Anand, D.; Monies, D.; Maddirevula, S.; Khan, A.O.; Algoufi, T.; Alowain, M.; Faqeih, E.; Alshammari, M.; Qudair, A.; et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Qual. Life Res. 2017, 136, 205–225. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2012, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagel, K.A.; Antaki, D.; Lian, A.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. Pathogenicity and functional impact of non-frameshifting insertion/deletion variation in the human genome. PLoS Comput. Biol. 2019, 15, e1007112. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Shiwang, L.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Hulo, N.; Bairoch, A.; Bulliard, V.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Pagni, M.; Sigrist, C.J. The PROSITE database. Nucleic Acids Res. 2006, 34, D227–D230. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nat. Cell Biol. 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rashed, M.S. Clinical applications of tandem mass spectrometry: Ten years of diagnosis and screening for inherited metabolic diseases. J. Chromatogr. B Biomed. Sci. Appl. 2001, 758, 27–48. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Almuhaizea, M.; Almass, R.; Alhargan, A.; Albader, A.; Salsench, E.M.; Howaidi, J.; Ihinger, J.; Karachunski, P.; Begtrup, A.; Castell, M.S.; et al. Truncating mutations in YIF1B cause a progressive encephalopathy with various degrees of mixed movement disorder, microcephaly, and epilepsy. Acta Neuropathol. 2020, 139, 791–794. [Google Scholar] [CrossRef] [Green Version]
- Chelban, V.; Alsagob, M.; Kloth, K.; Chirita-Emandi, A.; Vandrovcova, J.; Maroofian, R.; Davagnanam, I.; Bakhtiari, S.; AlSayed, M.D.; Rahbeeni, Z.; et al. Genetic and phenotypic characterization of NKX6-2 -related spastic ataxia and hypomyelination. Eur. J. Neurol. 2020, 27, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, L.; Lanko, K.; Alsagob, M.; Almass, R.; Al-Ahmadi, N.; Najafi, M.; Al-Muhaizea, M.; Alzaidan, H.; AlDhalaan, H.; Perenthaler, E.; et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain 2021, 144, 769–780. [Google Scholar] [CrossRef]
- Al-Muhaizea, M.A.; Aldeeb, H.; Almass, R.; Jaber, H.; Binhumaid, F.; Alquait, L.; Abukhalid, M.; Aldhalaan, H.; Alsagob, M.; Al-Bakheet, A.; et al. Genetics of ataxia telangiectasia in a highly consanguineous population. Ann. Hum. Genet. 2021, 86, 34–44. [Google Scholar] [CrossRef]
- Aldosary, M.; Baselm, S.; Abdulrahim, M.; Almass, R.; Alsagob, M.; AlMasseri, Z.; Huma, R.; AlQuait, L.; Al-Shidi, T.; Al-Obeid, E.; et al. SLC25A42 -associated mitochondrial encephalomyopathy: Report of additional founder cases and functional characterization of a novel deletion. JIMD Rep. 2021, 60, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B.; Kølvraa, S.; Christensen, E.; Kplvraa, S. Inhibition of the Glycine Cleavage System: Hyperglycinemia and Hyperglycinuria Caused by Valproic Acid. Epilepsia 1980, 21, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Watanabe, M.; Yoshitomi, H.; Yoshida, H.; Kimoto, H.; Kamiya, A.; Hayashi, T.; Akimura, T. Urinary Metabolites of Valproic Acid in Epileptic Patients. Biol. Pharm. Bull. 1998, 21, 304–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| This Study | Maddirevula et al., 2019 [7] | Patel et al., 2017 [8] | Alazami et al., 2015 [29] | ||||
|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
| Gender | F | M | F | NA | F | M | F |
| Age of onset | Few months | 1 month | 7 year | NA | 13 year | 1 year | 5 year |
| Consanguinity | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| Variant (cDNA) * | c.440A>G | c.440A>G | c.314C>T | c.314C>T | c.2452T>C | c.2452T>C | c.2452T>C |
| Variant (Protein) * | p.His147Arg | p.His147Arg | p.Pro105Leu | p.Pro105Leu | p.Trp818Arg | p.Trp818Arg | p.Trp818Arg |
| Variant Type | Missense | Missense | Missense | Missense | Missense | Missense | Missense |
| Seizure | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| GDD | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Cataract | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hearing impairment | Yes | NA | Yes | NA | NA | NA | NA |
| Microcephaly | Yes | Yes | NA | NA | Yes | Yes | Yes |
| Dolichocephaly | NA | Yes | NA | NA | NA | NA | NA |
| Scoliosis | NA | Yes | NA | NA | NA | NA | NA |
| Dysmorphic features | NA | High arched palate, low set ears, wide depressed nasal bridge, sialorrhea. | NA | NA | Peculiar orientation of alae nasae. | Micrognathia, small mandible, high arched palate, and small bell-shaped thoracic cage. HC = 37 cm (<3rd centile) | No |
| Motor difficulties | Yes | Yes | Started walking at the age of 2 years | NA | Yes | Yes | Yes |
| Renal dysfunction | NA | Glycine elevated | NA | NA | Yes | Yes | NA |
| Chest infections | No | Yes | NA | NA | Yes | Yes | NA |
| MRI | Medulla, pons, cerebellar vermis, and both cerebellar hemispheres were rather small in size, with enlarged cistern magnum and the box-like appearance of the fourth ventricle. | Generalized atrophy, septum pellucidum was patent and vermian hypoplasia | Small atrophic cerebellum with prominence of the posterior fossa CSF spaces | NA | Bilateral symmetrical alteration of the signal intensity of the white matter of both cerebral hemispheres in the form of bright signal intensity on T2W and flair images, a picture probably reflecting disturbed myelination | Markedly prominent cortical sulci and subarachnoid cisterns were noted | Hypomyelination and callosal thinning |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldhalaan, H.; AlBakheet, A.; AlRuways, S.; AlMutairi, N.; AlNakiyah, M.; AlGhofaili, R.; Cardona-Londoño, K.J.; Alahmadi, K.O.; AlQudairy, H.; AlRasheed, M.M.; et al. A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts. Genes 2022, 13, 92. https://doi.org/10.3390/genes13010092
Aldhalaan H, AlBakheet A, AlRuways S, AlMutairi N, AlNakiyah M, AlGhofaili R, Cardona-Londoño KJ, Alahmadi KO, AlQudairy H, AlRasheed MM, et al. A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts. Genes. 2022; 13(1):92. https://doi.org/10.3390/genes13010092
Chicago/Turabian StyleAldhalaan, Hesham, Albandary AlBakheet, Sarah AlRuways, Nouf AlMutairi, Maha AlNakiyah, Reema AlGhofaili, Kelly J. Cardona-Londoño, Khalid Omar Alahmadi, Hanan AlQudairy, Maha M. AlRasheed, and et al. 2022. "A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts" Genes 13, no. 1: 92. https://doi.org/10.3390/genes13010092
APA StyleAldhalaan, H., AlBakheet, A., AlRuways, S., AlMutairi, N., AlNakiyah, M., AlGhofaili, R., Cardona-Londoño, K. J., Alahmadi, K. O., AlQudairy, H., AlRasheed, M. M., Colak, D., Arold, S. T., & Kaya, N. (2022). A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts. Genes, 13(1), 92. https://doi.org/10.3390/genes13010092