

Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample, DNA Extraction, and Sequencing
2.2. Chloroplast Genome Assembly and Annotation
2.3. Sequence Analysis
2.4. Genome Comparison
2.5. Phylogenetic Reconstruction
3. Results and Discussion
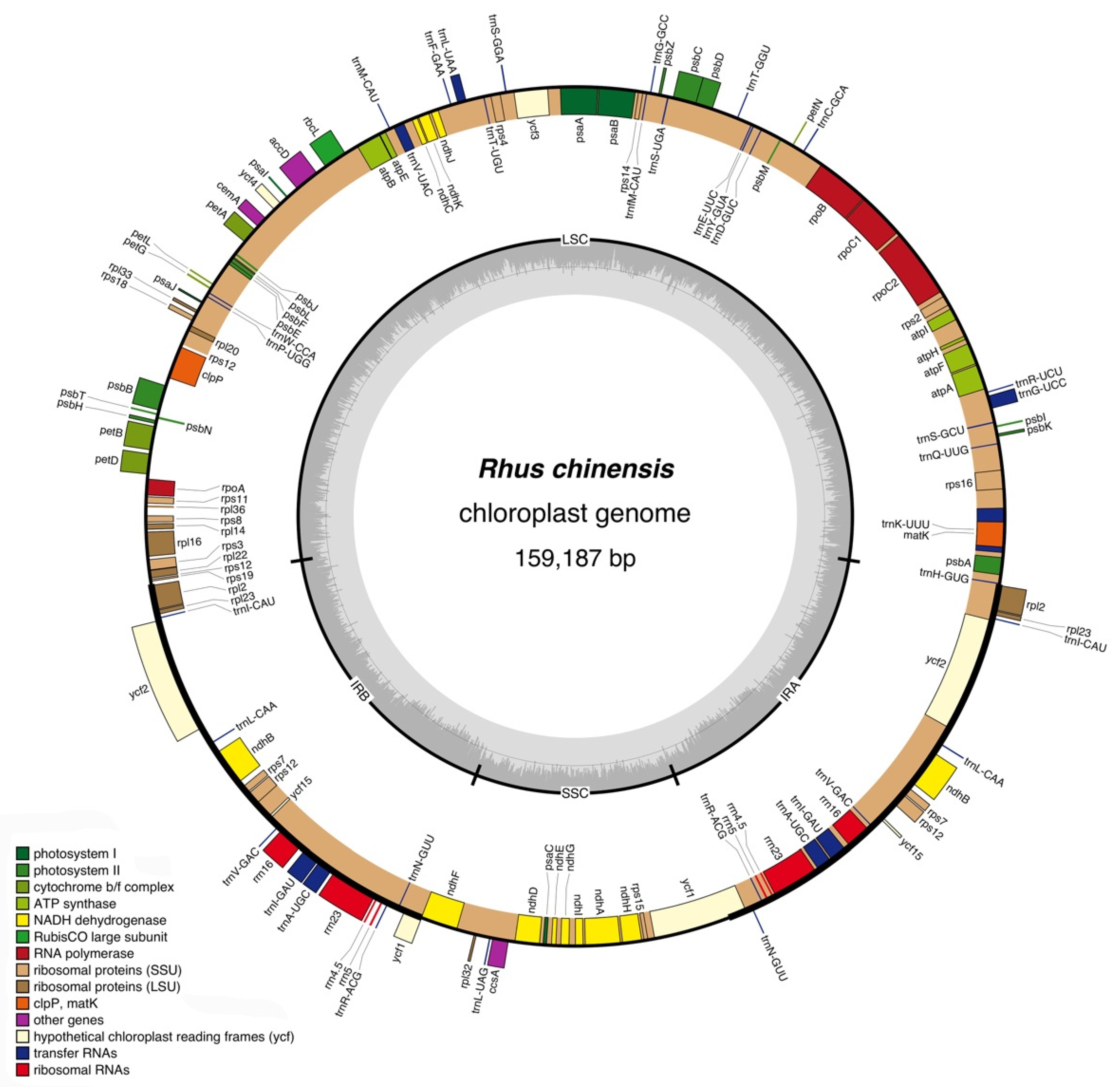
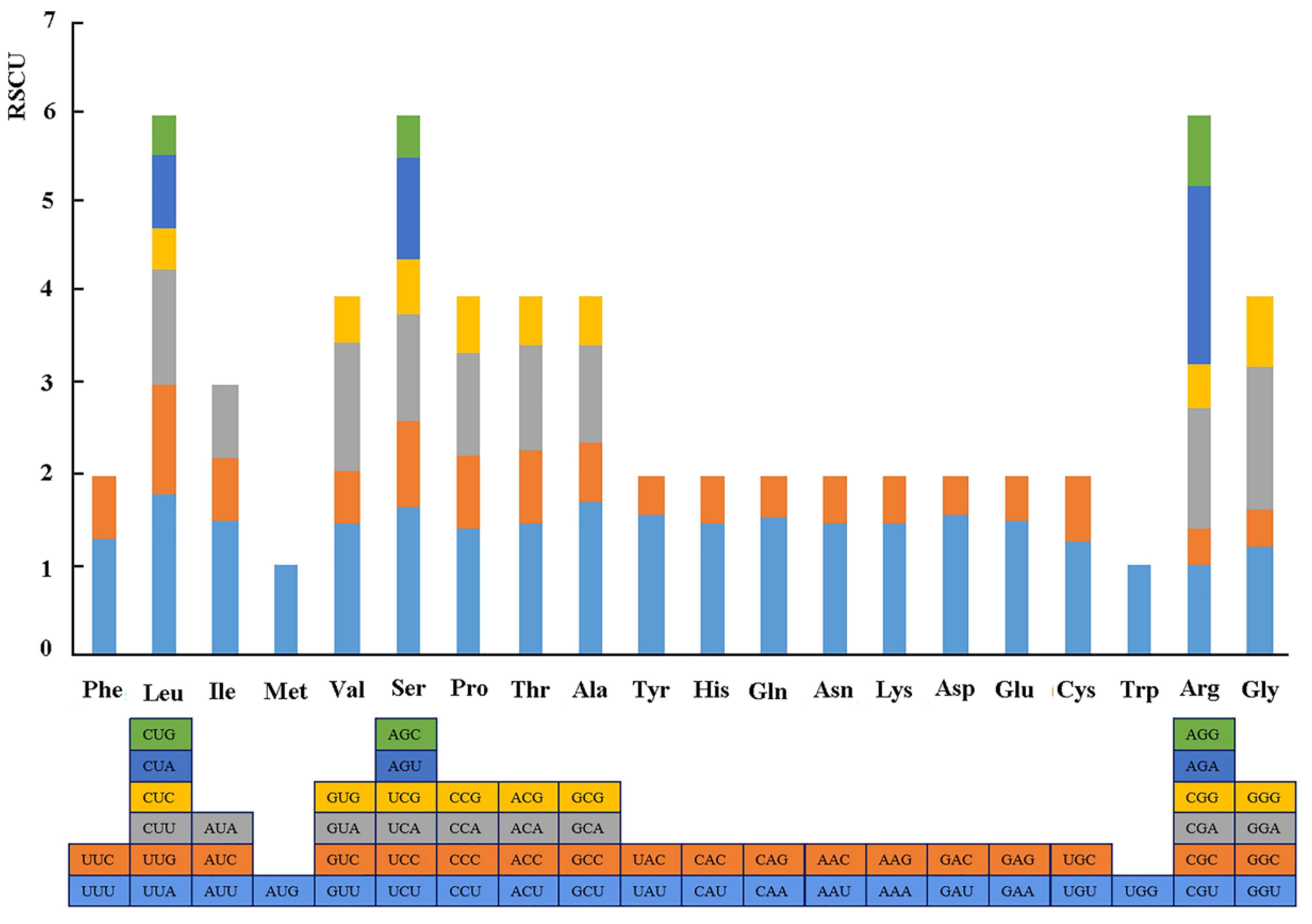
3.1. General Features of the Complete Chloroplast Genome of R. chinensis
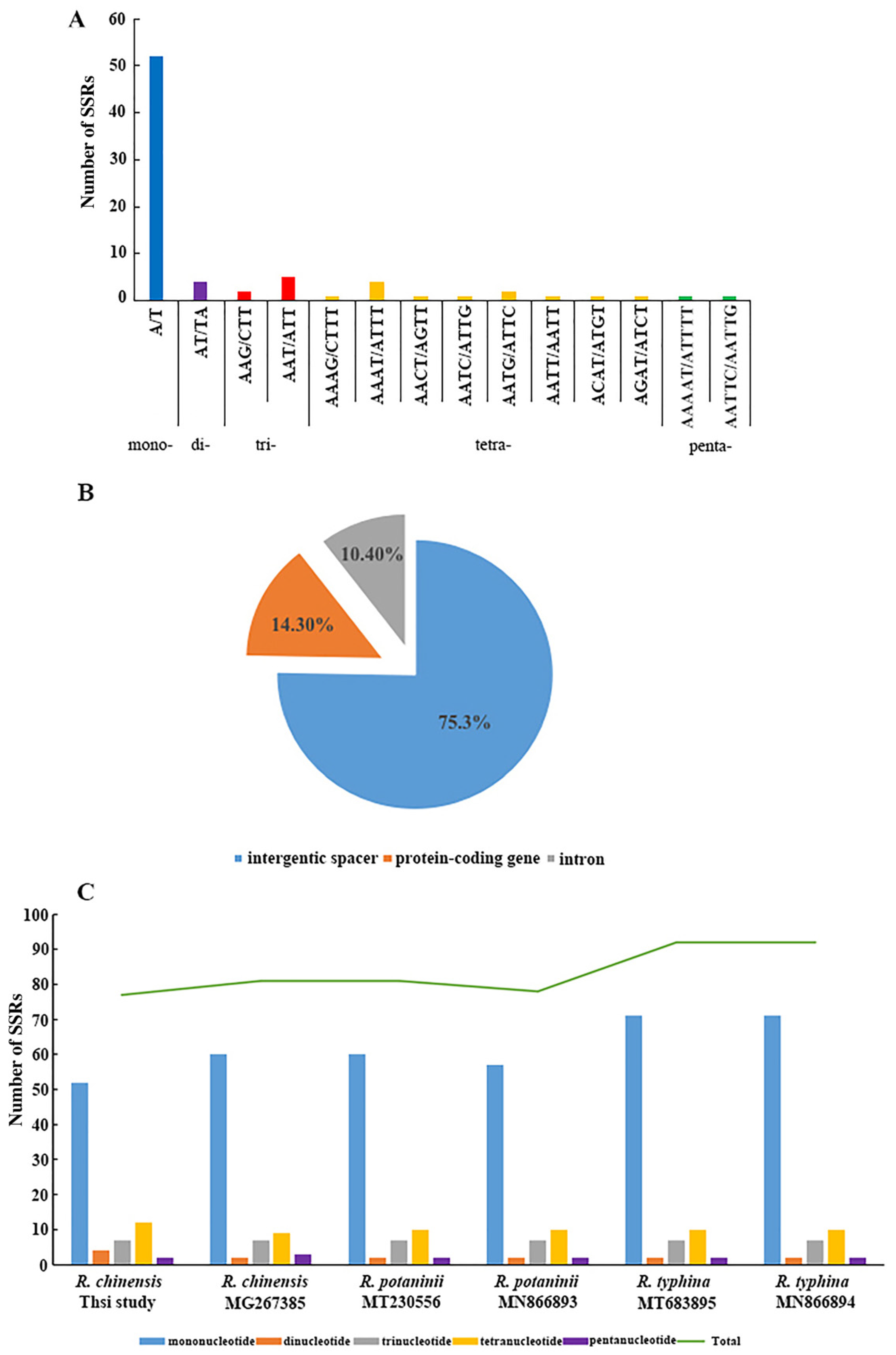
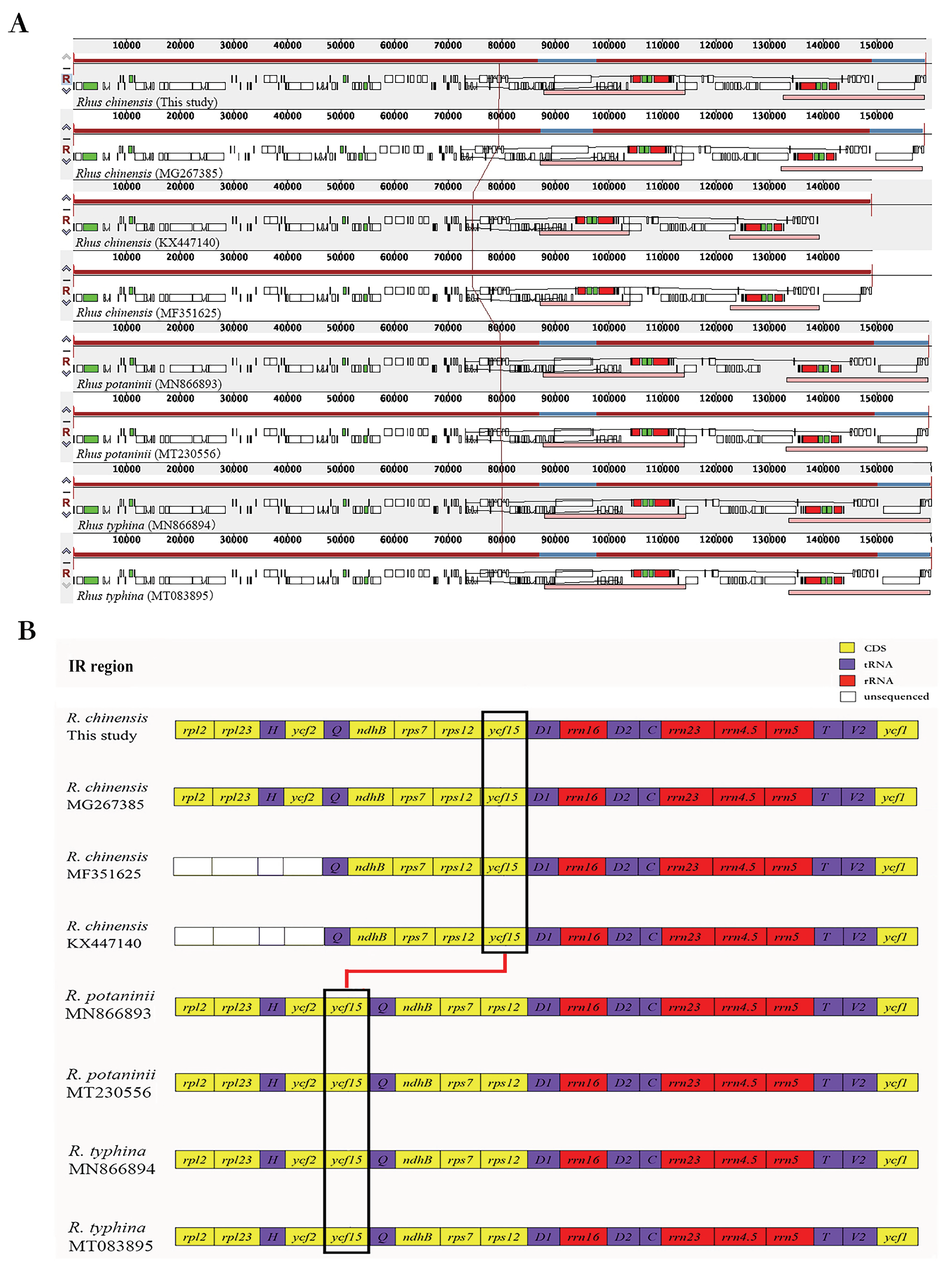
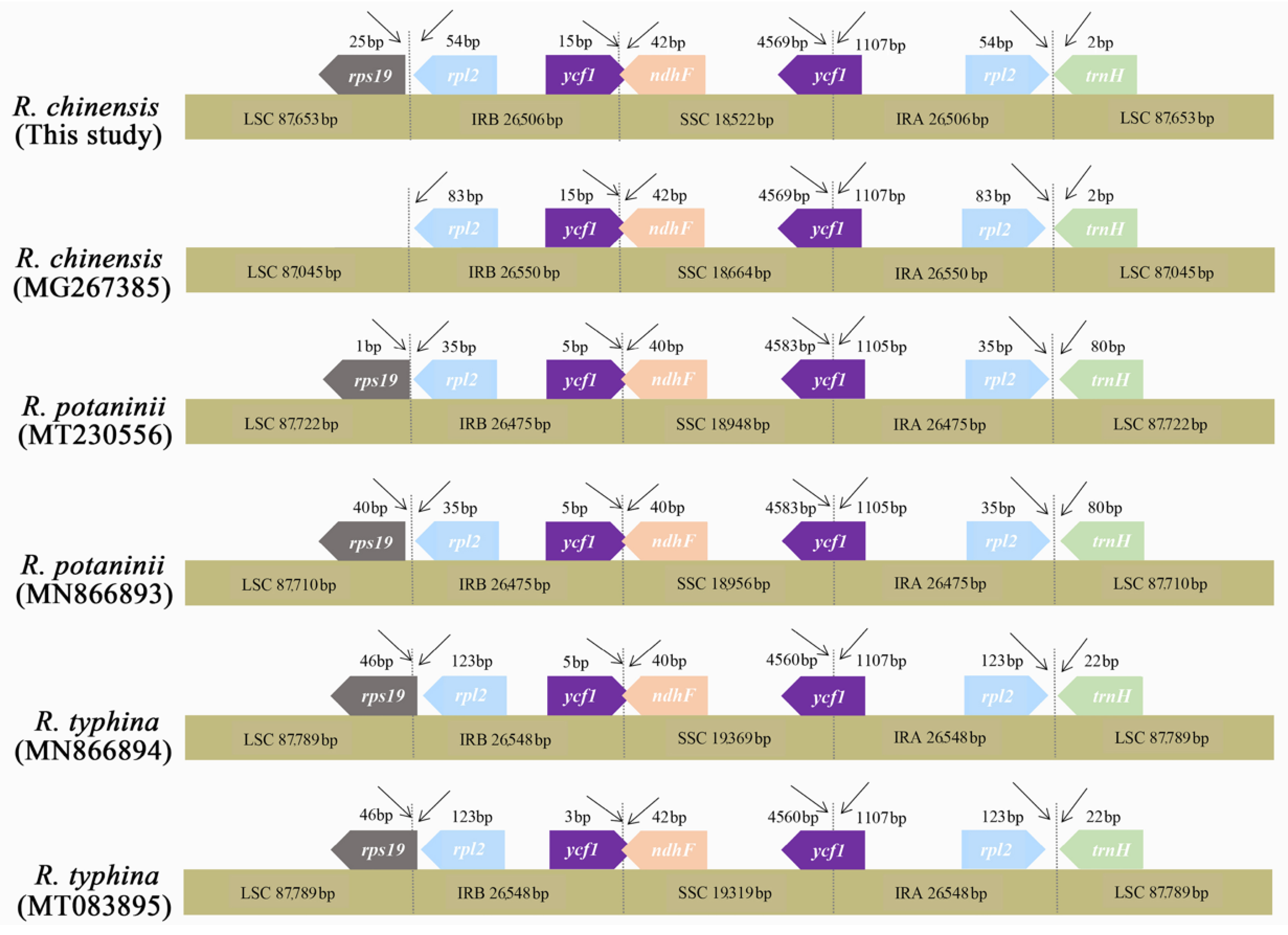
3.2. Comparison among R. chinensis Accessions
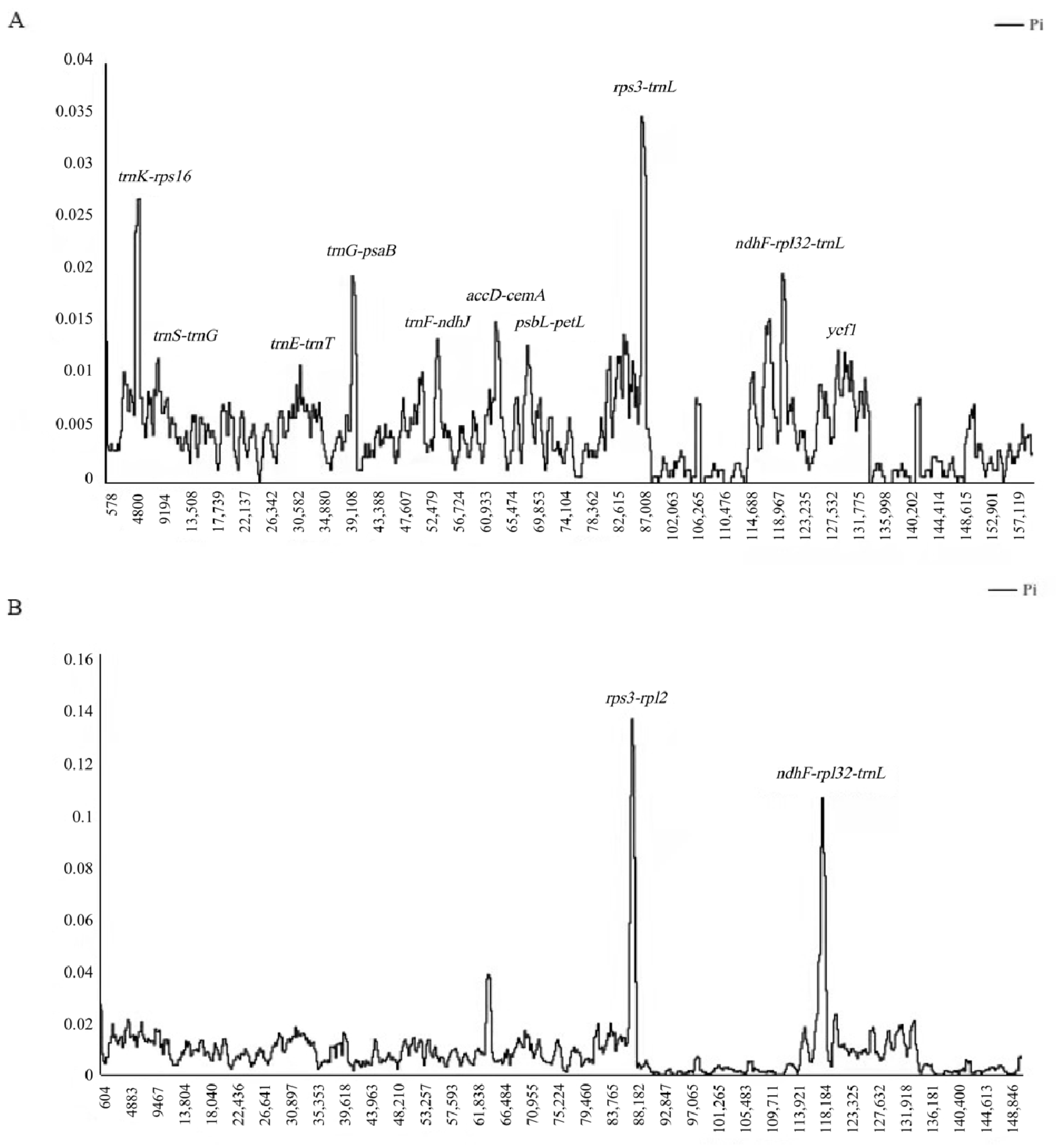
3.3. Comparison among the Rhus Species
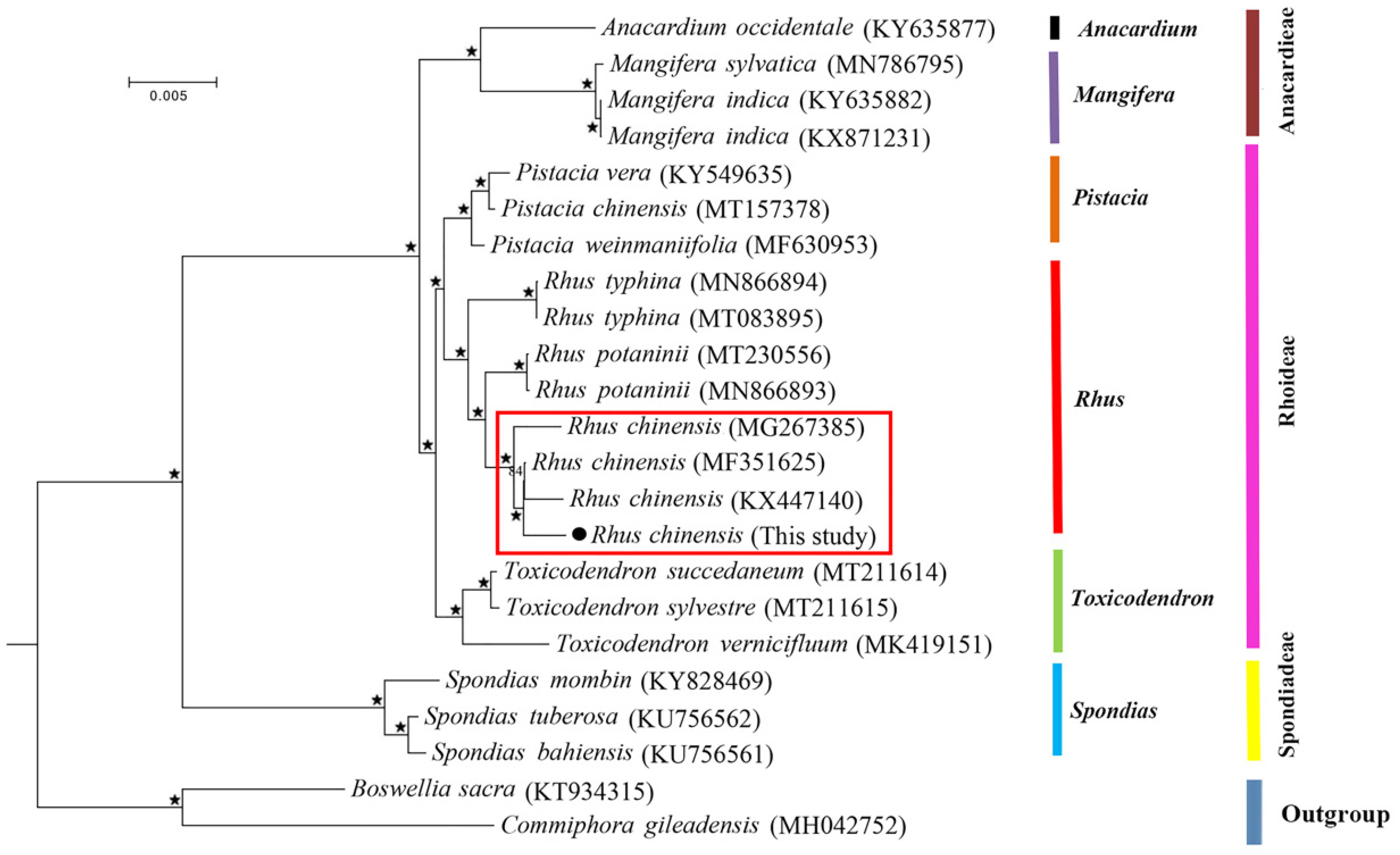
3.4. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rayne, S.; Mazza, G. Biological activities of extracts from sumac (Rhus spp.): A review. Plant Foods Hum. Nutr. 2007, 62, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkley, F.A. A monographic study of Rhus and its immediate allies in North and Central America, including the West Indies. Ann. Mo. Bot. Gard. 1937, 24, 265–498. [Google Scholar] [CrossRef]
- Andres-Hernandez, A.R.; Terrazas, T.; Salazar, G.; Ochoterena, H. Phylogenetic analysis based on structural and combined analyses of Rhus s.s. (Anacardiaceae). Bot. J. Linn. Soc. 2014, 176, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Barfod, M. Anacardiaceae. In Flora of China; Science Press: Beijing, China, 2008; p. 11. [Google Scholar]
- Chen, J.C.; Ho, T.Y.; Chang, Y.S.; Wu, S.L.; Li, C.C.; Hsiang, C.Y. Identification of Escherichia coli enterotoxin inhibitors from traditional medicinal herbs by in silico, in vitro, and in vivo analyses. J. Ethnopharmacol. 2009, 121, 372. [Google Scholar] [CrossRef] [PubMed]
- Djakpo, O.; Yao, W. Rhus chinensis and Galla chinensis—Folklore to modern evidence: Review. Phytother. Res. 2010, 24, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Oh, C.H. Growth and spatial distribution characteristics of Rhus javanica populations sowed on cut-slopes. J. Korean Soc. Environ. Restor. Technol. 2015, 18, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.J.; Yeo, W.J.; Choi, J.H.; Kim, N.C. Development of revegetation method using forest topsoils for ecological restoration of the slopes (I). J. Korean Soc. Environ. Restor. Technol. 2004, 7, 110–119. [Google Scholar]
- Zhang, G.X.; Qiao, G.X.; Zhong, T.S.; Zhang, W.Y. Fauna Sinica Insecta. Homoptera: Mindaridae and Pemphigidae; Science Press: Beijing, China, 1999; p. 14. [Google Scholar]
- Yang, Z.X.; Chen, X.M.; Havill, N.P.; Feng, Y.; Chen, H. Phylogeny of Rhus gall aphids (Hemiptera: Pemphigidae) based on combined molecular analysis of nuclear EF1 and mitochondrial COII genes. Entomol. Sci. 2010, 13, 351–357. [Google Scholar] [CrossRef]
- Su, T.C. Protection and utilization of wild Anacardiaceae plant resources in the mountainous areas of Eastern Liaoning. J. Liaoning For. Sci. Technol. 2016, 3, 59–60. [Google Scholar]
- Yang, X.Y.; Wang, J.; Li, C.; Ren, Z.M.; Ma, W.L. Cloning, expression and characterization of chalcone isomerase from medicinal plant Chinese sumac (Rhus chinensis). Chin. J. Chin. Mat. Med. 2019, 44, 3253–3260. [Google Scholar] [CrossRef]
- Ye, M.; Wen, X.; He, D.Q.; Wu, X.; Lai, W.F.; Zhang, X.Q.; Lin, Y.; Xu, W.; Li, X.W. Dammarane-type triterpenoids from the roots of Rhus chinensis and their preventive effects on zebrafish heart failure and thrombosis. J. Nat. Prod. 2020, 83, 362–373. [Google Scholar] [CrossRef]
- Ma, W.L.; Wu, M.; Wu, Y.; Ren, Z.M.; Zhong, Y. Cloning and characterisation of a phenylalanine ammonia-lyase gene from Rhus chinensis. Plant Cell. Rep. 2013, 32, 1179–1190. [Google Scholar] [CrossRef]
- Liang, Y.K.; Zhang, Y.; Wen, J.; Su, X.; Ren, Z.M. Evolutionary history of Rhus chinensis (Anacardiaceae) from the temperate and subtropical zones of China based on cpDNA and nuclear DNA sequences and ecological niche model. Front. Genet. 2019, 10, 171. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, Z.X.; Chen, X.M.; Liu, P.; Tang, Y.F. Effects of Chinese gallnut on photosynthetic characteristics and total nitrogen content of Rhus chinensis. Acta Ecol. Sin. 2013, 33, 6876–6884. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.K.; Kim, M.J.; Heo, K. Phylogeny of Korean Rhus spp. based on ITS and rbcL sequences. J. Korean Med. Sci. 2004, 12, 60–66. [Google Scholar]
- Miller, A.J.; Young, D.A.; Wen, J. Phylogeny and biogeography of Rhus (Anacardiaceae) based on ITS sequences data. Int. J. Plant Sci. 2001, 162, 1401–1407. [Google Scholar] [CrossRef]
- Yi, T.S.; Miller, A.J.; Wen, J. Phylogenetic and bio-geographic diversification of Rhus (Anacardiaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 2004, 33, 861–879. [Google Scholar] [CrossRef]
- Yi, T.S.; Miller, A.J.; Wen, J. Phylogeny of Rhus (Anacardiaceae) based on sequences of nuclear Nia-i3 intron and chloroplast trnC-trnD. Syst. Bot. 2007, 32, 379–391. [Google Scholar] [CrossRef]
- Kim, I.; Park, J.Y.; Lee, Y.S.; Joh, H.J.; Kang, S.J.; Murukarthick, J.M.; Lee, H.O.; Hur, Y.J.; Kim, Y.; Kim, K.H.; et al. The complete chloroplast genome sequence and intra-species diversity of Rhus chinensis. Plant. Breed. Biotechnol. 2017, 5, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, H.; Emes, M. No photosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Henry, R.J. Importance of plant diversity. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 1–5. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Cheng, Y.J.; De Vicente, M.C.; Meng, H.J.; Guo, W.W.; Tao, N.G.; Deng, X.X. A set of primers for analyzing chloroplast DNA diversity in Citrus and related genera. Tree Physiol. 2005, 25, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.N.; Zhu, Z.C.; Lu, Z.K.; Ding, Z.; Zhang, C.; Luan, F.S. The complete chloroplast genome sequence of the Sechium edule (Jacq.) Swartz. (Cucurbitaceae). Mitochondrial DNA Part B 2021, 6, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.X.; Movahedi, A.; Yang, W.G.; Xu, D.Z.; Jiang, C.B.; Xie, J.G.; Zhang, Y. The complete chloroplast genome and characteristics analysis of Musa basjoo Siebold. Mol. Biol. Rep. 2021, 48, 7113–7125. [Google Scholar] [CrossRef] [PubMed]
- Skuza, L.; Gastineau, R.; Sielska, A. The complete chloroplast genome of Secale sylvestre (Poaceae: Triticeae). J. Appl. Genet. 2022, 63, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, I.; Kim, J.K.; Park, J.Y.; Joh, H.J.; Park, H.S.; Lee, H.O.; Lee, S.C.; Hur, Y.J.; Yang, T.J. The complete chloroplast genome sequence of Rhus chinensis Mill (Anacardiaceae). Mitochondrial DNA Part B 2016, 1, 696–697. [Google Scholar] [CrossRef] [Green Version]
- Zuo, R.H.; Jiang, P.; Sun, C.B.; Chen, C.W.; Lou, X.J. Analysis of the chloroplast genome characteristics of Rhus chinensis by de novo sequencing. J. Biotechnol. 2020, 36, 772–781. [Google Scholar] [CrossRef]
- Zimmer, E.A.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects II next-gen approaches. J. Syst. Evol. 2015, 53, 371–379. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate denovo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef]
- Lohse, O.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef]
- MISA-Microsatellite Identification Tool. 2010. Available online: http://pgrc.ipk-gatersleben.de/misa/ (accessed on 27 September 2010).
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. Mafft online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Shigehiro, K.; Zmasek, C.M.; Osamu, N.; Kazutaka, K. Aleaves facilitates on-demand exploration of metazoan gene family trees on mafft sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Saina, J.K.; Li, Z.Z.; Gichira, A.W.; Liao, Y.Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima Mill) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Li, C.; Miao, H.M.; Xiong, S.J. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [Green Version]
- Goulding, S.E.; Wolfe, K.H.; Olmstead, R.G.; Morden, C.W. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.C.; Zhang, Y.Z.; Geng, H.M.; Chen, S.L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F. Plastid genomes of the North American Rhus integrifolia-ovata complex and phylogenomic implications of inverted repeat structural evolution in Rhus L. PeerJ 2020, 8, e9315. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S.L. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.P.; Xu, C.; Li, D.L.; Jin, X.B.; Lu, Q.; Suo, Z.L. Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 2016, 4, e2699. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Panesar, P.S.; Bera, M.B.; Kaur, V. Simple sequence repeat markers in genetic divergence and marker-assisted selection of rice cultivars: A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 41–49. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Chaney, L.; Mangelson, R.; Ramaraj, T.; Jellen, E.N.; Maughan, P.J. The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Appl. Plant Sci. 2016, 4, 1600063. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [Green Version]
- Dobes, C.; Paule, J. A comprehensive chloroplast DNA-based phylogeny of the genus Potentilla (Rosaceae): Implications for its geographic origin, phylogeography and generic circumscription. Mol. Phylogenet. Evol. 2010, 56, 156–175. [Google Scholar] [CrossRef]
- Pan, Y.J.; Feng, J.L.; Nie, L.P.; Cui, Y.X.; Yang, C.H.; Lin, Y.; Yao, H. The complete chloroplast genome sequence of Rhus potaninii. Mitochondrial DNA Part B 2020, 3, 2425–2426. [Google Scholar] [CrossRef]
- Sun, L.; Fang, L.; Zhang, Z.; Chang, X.; Penny, D.; Zhong, B. Chloroplast phylogenomic inference of green algae relationships. Sci. Rep. 2016, 6, 20528. [Google Scholar] [CrossRef]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Family | Accession No. | Size (bp) |
|---|---|---|---|
| R. chinensis | Anacardiaceae | This Study | 159,187 |
| R. chinensis | Anacardiaceae | KX447140 | 149,011 |
| R. chinensis | Anacardiaceae | MF351625 | 149,094 |
| R. chinensis | Anacardiaceae | MG267385 | 158,809 |
| R. potaninii | Anacardiaceae | MT230556 | 159,620 |
| R. potaninii | Anacardiaceae | MN866893 | 159,616 |
| R. typhina | Anacardiaceae | MT083895 | 160,204 |
| R. typhina | Anacardiaceae | MN866894 | 160,254 |
| Pistacia chinensis | Anacardiaceae | MT157378 | 160,596 |
| Pistacia vera | Anacardiaceae | KY549635 | 160,674 |
| P. weinmanniifolia | Anacardiaceae | MF630953 | 160,767 |
| Toxicodendron succedaneum | Anacardiaceae | MT211614 | 150,710 |
| Toxicodendron sylvestre | Anacardiaceae | MT211615 | 159,600 |
| Toxicodendron vernicifluum | Anacardiaceae | MK419151 | 159,571 |
| Mangifera indica | Anacardiaceae | KX871231 | 157,780 |
| Mangifera indica | Anacardiaceae | KY635882 | 157,780 |
| Mangifera sylvatica | Anacardiaceae | MN786795 | 158,106 |
| Anacardium occidentale | Anacardiaceae | KY635877 | 172,199 |
| Spondias tuberosa | Anacardiaceae | KU756562 | 162,039 |
| Spondias bahiensis | Anacardiaceae | KU756561 | 162,218 |
| Spondias mombin | Anacardiaceae | KY828469 | 162,302 |
| B. sacra | Burseraceae | KT934315 | 160,543 |
| C. gileadensis | Burseraceae | MH042752 | 160,268 |
| Species | R. chinensis | R. potaninii | R. typhina | |||||
|---|---|---|---|---|---|---|---|---|
| Accession No. | OP326720 | KX447140 | MF351625 | MG267385 | MN866893 | MT230556 | MN866894 | MT083895 |
| Location | Hubei | Gangwon | Shandong | Anhui | Beijing | Shaanxi | Unknown | Shandong |
| Size (bp) | 159,187 | 149,011 | 149,094 | 158,809 | 159,616 | 159,620 | 160,254 | 160,204 |
| LSC (bp) | 87,653 | 96,882 | 97,246 | 87,045 | 87,710 | 87,722 | 87,789 | 87,789 |
| SSC (bp) | 18,522 | 18,674 | 18,644 | 18,664 | 18,956 | 18,948 | 19,453 | 19,319 |
| IR (bp) | 26,506 | 16,741 | 16,602 | 26,550 | 26,475 | 26,475 | 26,506 | 26,548 |
| No. of total genes | 132 | 126 | 126 | 130 | 131 | 133 | 130 | 130 |
| Protein-coding genes | 86 | 82 | 82 | 85 | 86 | 86 | 85 | 85 |
| tRNAs | 37 | 36 | 36 | 37 | 37 | 37 | 37 | 37 |
| rRNAs | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Overall GC content (%) | 37.8 | 37.8 | 37.9 | 37.9 | 37.9 | 37.9 | 37.8 | 37.8 |
| GC content of LSC (%) | 35.9 | 36.2 | 36.2 | 36.0 | 36.0 | 36.0 | 35.8 | 35.8 |
| GC content of SSC (%) | 32.6 | 32.7 | 32.7 | 32.6 | 32.6 | 32.6 | 32.5 | 32.5 |
| GC content of IR (%) | 42.9 | 45.4 | 45.5 | 42.6 | 43.0 | 43.0 | 43.0 | 43.0 |
| Gene | Exon Ⅰ (bp) | Intron Ⅰ (bp) | Exon Ⅱ (bp) | Intron Ⅱ (bp) | Exon Ⅲ (bp) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| rps16 | 226 | 226 | 227 | 226 | 894 | 899 | 896 | 882 | 38 | 38 | 40 | 38 | ||||||||
| atpF | 411 | 748 | 748 | 760 | 747 | 156 | 156 | 144 | 156 | |||||||||||
| rpoC1 | 1626 | 760 | 760 | 760 | 755 | 435 | ||||||||||||||
| ycf3 | 155 | 813 | 797 | 797 | 798 | 226 | 732 | 735 | 733 | 734 | 126 | |||||||||
| clpP | 228 | 631 | 653 | 638 | 637 | 291 | 291 | 292 | 291 | 773 | 776 | 768 | 775 | 69 | 69 | 71 | 69 | |||
| petB | 6 | 785 | 786 | 785 | 772 | 642 | ||||||||||||||
| petD | 8 | 741 | 724 | 741 | 750 | 475 | ||||||||||||||
| rpl16 | 402 | 1068 | 1071 | 1068 | 1133 | 9 | ||||||||||||||
| rpl2 | 393 | 662 | 662 | 662 | 661 | 435 | ||||||||||||||
| ndhB | 777 | 681 | 756 | |||||||||||||||||
| rps12 | 232 | 536 | 26 | |||||||||||||||||
| ndhA | 541 | 1120 | 1120 | 1129 | 1124 | 467 | 467 | 551 | 467 | |||||||||||
| rps12 | 26 | 536 | 232 | |||||||||||||||||
| ndhB | 756 | 681 | 777 | |||||||||||||||||
| rpl2 | 434 | — | — | 434 | 665 | — | — | 664 | 391 | — | — | 391 | ||||||||
| trnK-UUU | 35 | 2599 | 2593 | 2596 | 2596 | 37 | ||||||||||||||
| trnG-UCC | 23 | 714 | 713 | 713 | 713 | 47 | ||||||||||||||
| trnL-UAA | 37 | 450 | 50 | |||||||||||||||||
| trnV-UAC | 37 | 585 | 39 | |||||||||||||||||
| trnA-UGC | 35 | 841 | 38 | |||||||||||||||||
| trnI-GAU | 35 | 950 | 949 | 950 | 940 | 42 | ||||||||||||||
| trnI-GAU | 42 | 950 | 949 | 950 | 940 | 35 | ||||||||||||||
| trnA-UGC | 38 | 841 | 35 | |||||||||||||||||
| Species | Region | Total Sites | Variable Sites | Informative Sites | Nucleotide Diversity |
|---|---|---|---|---|---|
| R. chinensis | Large single-copy region | 98,636 | 969 | 29 | 0.00584 |
| Small single-copy region | 18,751 | 281 | 20 | 0.00783 | |
| Inverted repeat region | 27,478 | 48 | 11 | 0.00156 | |
| Complete cp genome | 160,884 | 1436 | 93 | 0.00502 | |
| Six Rhus species | Large single-copy region | 89,280 | 1743 | 1075 | 0.00958 |
| Small single-copy region | 19,572 | 616 | 435 | 0.01614 | |
| Inverted repeat region | 27,432 | 109 | 61 | 0.00184 | |
| Complete cp genome | 162,196 | 2931 | 1621 | 0.00841 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wen, J.; Su, X.; Ren, Z. Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis. Genes 2022, 13, 1936. https://doi.org/10.3390/genes13111936
Xu Y, Wen J, Su X, Ren Z. Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis. Genes. 2022; 13(11):1936. https://doi.org/10.3390/genes13111936
Chicago/Turabian StyleXu, Yujie, Jun Wen, Xu Su, and Zhumei Ren. 2022. "Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis" Genes 13, no. 11: 1936. https://doi.org/10.3390/genes13111936
APA StyleXu, Y., Wen, J., Su, X., & Ren, Z. (2022). Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis. Genes, 13(11), 1936. https://doi.org/10.3390/genes13111936