
Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype–Phenotype Correlation
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
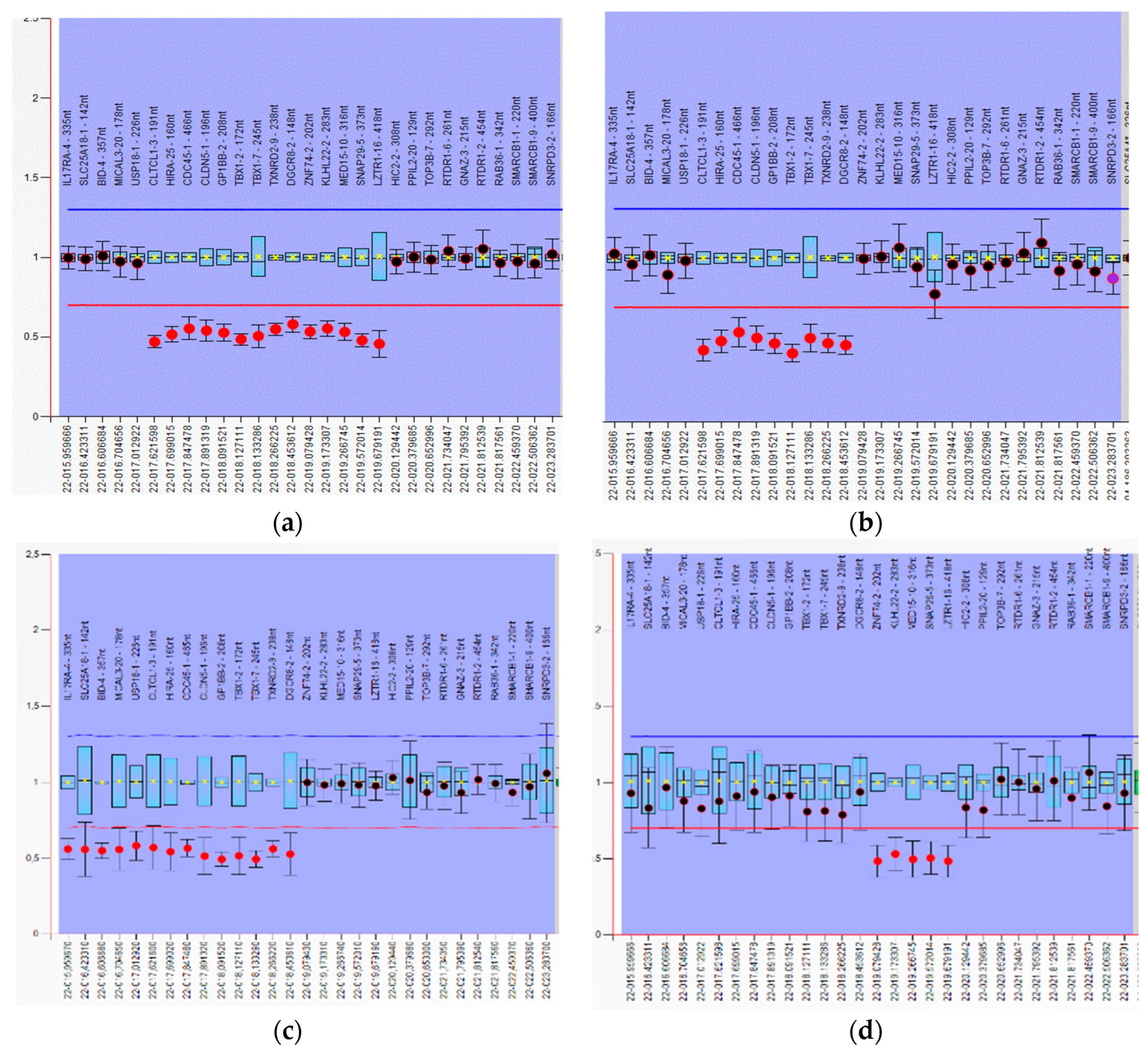
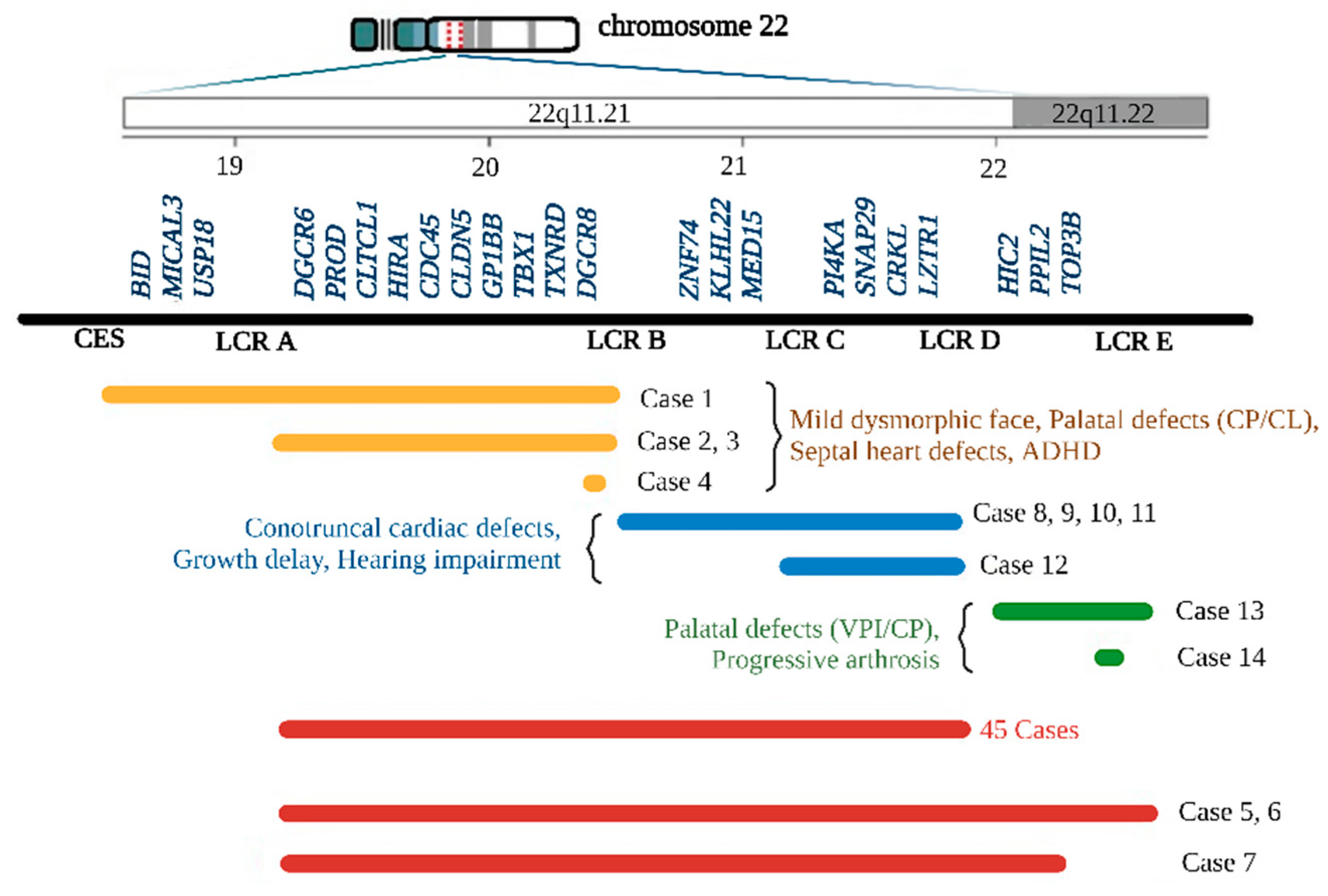
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Goldenberg, P. An Update on Common Chromosome Microdeletion and Microduplication Syndromes. Pediatr. Ann. 2018, 47, e198–e203. [Google Scholar] [CrossRef] [PubMed]
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech Lang Pathol. 2019, 28, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Jackson, O.; Crowley, T.B.; Sharkus, R.; Smith, R.; Jeong, S.; Solot, C.; McDonald-Mcginn, D. Palatal evaluation and treatment in 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2019, 179, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. A 2018, 176, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevado, J.; Mergener, R.; Palomares-Bralo, M.; Souza, K.R.; Vallespin, E.; Mena, R.; Martinez-Glez, V.; Mori, M.A.; Santos, F.; Garcia-Minaur, S.; et al. New microdeletion and microduplication syndromes: A comprehensive review. Genet. Mol. Biol. 2014, 37, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacihamdioglu, B.; Hacihamdioglu, D.; Delil, K. 22q11 deletion syndrome: Current perspective. Appl. Clin. Genet. 2015, 8, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Yu, S.; Newkirk, H.; Kibiryeva, N.; Holt, A.; Butler, M.G. Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet Genome Res. 2009, 124, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Delio, M.; Haque, N.; Castellanos, R.; Hestand, M.S.; Vermeesch, J.R.; Morrow, B.E.; Zheng, D. Variant discovery and breakpoint region prediction for studying the human 22q11.2 deletion using BAC clone and whole genome sequencing analysis. Hum. Mol. Genet. 2016, 25, 3754–3767. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Michaelovsky, E.; Frisch, A.; Carmel, M.; Patya, M.; Zarchi, O.; Green, T.; Basel-Vanagaite, L.; Weizman, A.; Gothelf, D. Genotype-phenotype correlation in 22q11.2 deletion syndrome. BMC Med. Genet. 2012, 13, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald-McGinn, D.M.; Zackai, E.H. Genetic counseling for the 22q11.2 deletion. Dev. Disabil. Res. Rev. 2008, 14, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Crowley, B.; Ruffner, M.; McDonald McGinn, D.M.; Sullivan, K.E. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2018, 176, 2082–2086. [Google Scholar] [CrossRef]
- Vorstman, J.A.; Jalali, G.R.; Rappaport, E.F.; Hacker, A.M.; Scott, C.; Emanuel, B.S. MLPA: A rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum. Mutat. 2006, 27, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzaru, M.; Rusu, C.; Volosciuc, M.; Braha, E.; Butnariu, L.; Ivanov, I.; Gramescu, M.; Popescu, R.; Caba, L.; Sireteanu, A.; et al. Improvement of genetic diagnostic strategy in velo-cardio-facial syndrome. Rev. Med. Chir. Soc. Med. Nat. Iasi 2011, 115, 756–761. [Google Scholar] [PubMed]
- Carli, D.; Moroni, A.; Eleonora, D.G.; Zonta, A.; Montin, D.; Licciardi, F.; Aidala, E.; Bordese, R.; Carlo, P.N.; Brusco, A.; et al. Atypical microdeletion 22q11.2 in a patient with tetralogy of Fallot. J. Genet. 2021, 100, 5. [Google Scholar] [CrossRef]
- Burnside, R.D. 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet Genome Res. 2015, 146, 89–99. [Google Scholar] [CrossRef]
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-Number Variation of the Glucose Transporter Gene SLC2A3 and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 96, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergaelen, E.; Swillen, A.; Van Esch, H.; Claes, S.; Van Goethem, G.; Devriendt, K. 3 generation pedigree with paternal transmission of the 22q11.2 deletion syndrome: Intrafamilial phenotypic variability. Eur. J. Med. Genet. 2015, 58, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Rump, P.; de Leeuw, N.; van Essen, A.J.; Verschuuren-Bemelmans, C.C.; Veenstra-Knol, H.E.; Swinkels, M.E.; Oostdijk, W.; Ruivenkamp, C.; Reardon, W.; de Munnik, S.; et al. Central 22q11.2 deletions. Am. J. Med. Genet. A 2014, 164A, 2707–2723. [Google Scholar] [CrossRef]
- Clements, C.C.; Wenger, T.L.; Zoltowski, A.R.; Bertollo, J.R.; Miller, J.S.; de Marchena, A.B.; Mitteer, L.M.; Carey, J.C.; Yerys, B.E.; Zackai, E.H.; et al. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol. Autism. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racedo, S.E.; McDonald-McGinn, D.M.; Chung, J.H.; Goldmuntz, E.; Zackai, E.; Emanuel, B.S.; Zhou, B.; Funke, B.; Morrow, B.E. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am. J. Hum. Genet. 2015, 96, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindewolf, E.; Khalek, N.; Johnson, M.P.; Gebb, J.; Coleman, B.; Crowley, T.B.; Zackai, E.H.; McDonald-McGinn, D.M.; Moldenhauer, J.S. Expanding the fetal phenotype: Prenatal sonographic findings and perinatal outcomes in a cohort of patients with a confirmed 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2018, 176, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- D’Angelo, C.S.; Jehee, F.S.; Koiffmann, C.P. An inherited atypical 1 Mb 22q11.2 deletion within the DGS/VCFS 3 Mb region in a child with obesity and aggressive behavior. Am. J. Med. Genet. A 2007, 143A, 1928–1932. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Totten, A.H.; Elkahloun, A.G.; Johnson, K.R.; Hurst, A.C.; Goldman, F.; Groves, A.K.; Mikhail, F.M.; Atkinson, T.P. Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J. Allergy Clin. Immunol. 2020, 145, 358–367.e352. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Y.; Collins, A.; James, P.A.; McGillivray, G.; Stark, Z.; Gordon, C.T.; Leventer, R.J.; Pope, K.; Forbes, R.; Crolla, J.A.; et al. Phenotypic variability of distal 22q11.2 copy number abnormalities. Am. J. Med. Genet. A 2011, 155A, 1623–1633. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Ou, Z.; Shaw, C.A.; Belmont, J.W.; Patel, M.S.; Hummel, M.; Amato, S.; Tartaglia, N.; Berg, J.; Sutton, V.R.; et al. 22q11.2 distal deletion: A recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am. J. Hum. Genet. 2008, 82, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Karbarz, M. Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels. Genes 2020, 11, 977. [Google Scholar] [CrossRef]
- Bassett, A.S.; Chow, E.W.; Husted, J.; Hodgkinson, K.A.; Oechslin, E.; Harris, L.; Silversides, C. Premature death in adults with 22q11.2 deletion syndrome. J. Med. Genet. 2009, 46, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzaru, M.; Rusu, C.; Voloşciuc, M.; Braha, E.; Butnariu, L.; Gramescu¹, M.; R, P.; Caba, L.; Bujoran, C.; Ivanov, I.; et al. Benefits of cytogenetic testing in diagnosis of plurimalformative syndromes with congenital heart defects. Rev. Romana Med. Lab. 2012, 20, 265–272. [Google Scholar]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Gao, W.; Higaki, T.; Eguchi-Ishimae, M.; Iwabuki, H.; Wu, Z.; Yamamoto, E.; Takata, H.; Ohta, M.; Imoto, I.; Ishii, E.; et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Hum. Genome Var. 2015, 2, 15004. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef]
- Moon, A.M.; Guris, D.L.; Seo, J.H.; Li, L.; Hammond, J.; Talbot, A.; Imamoto, A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev. Cell 2006, 10, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Rozas, M.F.; Benavides, F.; Leon, L.; Repetto, G.M. Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: Systematic review and meta-analysis. Orphanet J. Rare Dis. 2019, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.S.; Genovese, A.; Butler, M.G. Deletion of TOP3B Is Associated with Cognitive Impairment and Facial Dysmorphism. Cytogenet Genome Res. 2016, 150, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Spineli-Silva, S.; Bispo, L.M.; Gil-da-Silva-Lopes, V.L.; Vieira, T.P. Distal deletion at 22q11.2 as differential diagnosis in Craniofacial Microsomia: Case report and literature review. Eur. J. Med. Genet. 2018, 61, 262–268. [Google Scholar] [CrossRef]
- Swillen, A.; McDonald-McGinn, D. Developmental trajectories in 22q11.2 deletion. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Swillen, A.; Moss, E.; Duijff, S. Neurodevelopmental outcome in 22q11.2 deletion syndrome and management. Am. J. Med. Genet. A 2018, 176, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Sireteanu, A.; Popescu, R.; Braha, E.E.; Bujoran, C.; Butnariu, L.; Caba, L.; Graur, E.; Gorduza, E.V.; Grămescu, M.; Ivanov, I.C.; et al. Detection of chromosomal imbalances using combined MLPA kits in patients with syndromic intellectual disability. Rev. Romana Med. Lab. 2014, 22, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Zinkstok, J.R.; Boot, E.; Bassett, A.S.; Hiroi, N.; Butcher, N.J.; Vingerhoets, C.; Vorstman, J.A.S.; van Amelsvoort, T. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatry 2019, 6, 951–960. [Google Scholar] [CrossRef]
- Cancrini, C.; Puliafito, P.; Digilio, M.C.; Soresina, A.; Martino, S.; Rondelli, R.; Consolini, R.; Ruga, E.M.; Cardinale, F.; Finocchi, A.; et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J. Pediatr. 2014, 164, 1475–1480.e1472. [Google Scholar] [CrossRef]
- Provenzani, U.; Damiani, S.; Bersano, I.; Singh, S.; Moschillo, A.; Accinni, T.; Brondino, N.; Oliver, D.; Fusar-Poli, P. Prevalence and incidence of psychotic disorders in 22q11.2 deletion syndrome: A meta-analysis. Int. Rev. Psychiatry 2022, 1–13. [Google Scholar] [CrossRef]
- Woodward, K.J.; Stampalia, J.; Vanyai, H.; Rijhumal, H.; Potts, K.; Taylor, F.; Peverall, J.; Grumball, T.; Sivamoorthy, S.; Alinejad-Rokny, H.; et al. Atypical nested 22q11.2 duplications between LCR22B and LCR22D are associated with neurodevelopmental phenotypes including autism spectrum disorder with incomplete penetrance. Mol. Genet. Genomic. Med. 2019, 7, e00507. [Google Scholar] [CrossRef] [Green Version]
- Van Batavia, J.P.; Crowley, T.B.; Burrows, E.; Zackai, E.H.; Sanna-Cherchi, S.; McDonald-McGinn, D.M.; Kolon, T.F. Anomalies of the genitourinary tract in children with 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2019, 179, 381–385. [Google Scholar] [CrossRef]
- Sanna-Cherchi, S.; Kiryluk, K.; Burgess, K.E.; Bodria, M.; Sampson, M.G.; Hadley, D.; Nees, S.N.; Verbitsky, M.; Perry, B.J.; Sterken, R.; et al. Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 2012, 91, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Bartram, M.P.; Dafinger, C.; Habbig, S.; Benzing, T.; Schermer, B.; Muller, R.U. Loss of Dgcr8-mediated microRNA expression in the kidney results in hydronephrosis and renal malformation. BMC Nephrol. 2015, 16, 55. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol. Rev. 2019, 287, 186–201. [Google Scholar] [CrossRef]
- Sullivan, K.E. Immune Biomarkers of Autoimmunity in Chromosome 22q11.2 Deletion Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 2377–2378. [Google Scholar] [CrossRef] [PubMed]
- Verheij, E.; Derks, L.S.M.; Stegeman, I.; Thomeer, H. Prevalence of hearing loss and clinical otologic manifestations in patients with 22q11.2 deletion syndrome: A literature review. Clin. Otolaryngol. 2017, 42, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Nehme, R.; Pietilainen, O.; Artomov, M.; Tegtmeyer, M.; Valakh, V.; Lehtonen, L.; Bell, C.; Singh, T.; Trehan, A.; Sherwood, J.; et al. The 22q11.2 region regulates presynaptic gene-products linked to schizophrenia. Nat. Commun. 2022, 13, 3690. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.; Papolos, D.; Pandita, R.K.; Faedda, G.L.; Veit, S.; Goldberg, R.; Shprintzen, R.; Kucherlapati, R.; Morrow, B. Molecular analysis of velo-cardio-facial syndrome patients with psychiatric disorders. Am. J. Hum. Genet. 1997, 60, 851–859. [Google Scholar] [PubMed]
- Xue, J.; Shen, R.; Xie, M.; Liu, Y.; Zhang, Y.; Gong, L.; Li, H. 22q11.2 recurrent copy number variation-related syndrome: A retrospective analysis of our own microarray cohort and a systematic clinical overview of ClinGen curation. Transl. Pediatr. 2021, 10, 3273–3281. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phenotypic Features | No. of Cases | F | M | |
|---|---|---|---|---|
| Dysmorphism | Characteristic | 32 | 14 | 18 |
| Mild | 7 | 2 | 5 | |
| Atypic | 1 | - | 1 | |
| Heart defects | Tetralogy of Fallot | 20 | 6 | 14 |
| Ventricular septal defect | 15 | 5 | 10 | |
| Truncus arteriosus | 4 | 1 | 3 | |
| Aortic arch anomalies | 3 | - | 3 | |
| Pulmonary atresia | 3 | - | 3 | |
| Atrial septal defect | 2 | - | 2 | |
| Patent ductus arteriosus | 1 | - | 1 | |
| Pulmonary stenosis | 1 | 1 | - | |
| Interrupted aortic arch | 1 | - | 1 | |
| Bicuspid aortic valve | 1 | - | 1 | |
| Other | 4 | 2 | 2 | |
| Palatal defects | Velopharyngeal incompetence | 20 | 11 | 9 |
| Dysphagia | 8 | 2 | 6 | |
| Cleft Palate | 5 | 1 | 4 | |
| Submucosal cleft palate | 2 | - | 2 | |
| Cleft lip and palate | 1 | 1 | - | |
| Development delay ± cognitive impairment | Mild | 14 | 6 | 8 |
| Moderate | 8 | 3 | 5 | |
| Severe | 5 | 2 | 3 | |
| Psychiatric/Behavior problems | Attention deficit | 7 | 5 | 2 |
| Autism-spectrum disorders | 2 | 1 | 1 | |
| Schizophrenia | 1 | 1 | - | |
| Hearing impairment | 5 | 3 | 2 | |
| Growth delay | 18 | 5 | 13 | |
| Hypotonia | 2 | - | 2 | |
| Hypocalcemia | 21 | 10 | 11 | |
| Seizures | 16 | 9 | 7 | |
| Immunodeficiency | 21 | 10 | 11 | |
| Urogenital anomaly | Hydronephrosis, | 7 | 5 | 2 |
| Cryptorchidism | 3 | - | 3 | |
| Renal hypoplasia | 2 | 1 | 1 | |
| Multicystic dysplastic kidney | 1 | 1 | - | |
| Hypospadias | 1 | - | 1 | |
| Case No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Deletion type | CES to B | A to B | A to B | DGCR8 | A to E | A to E | A to D+ | B to D | B to D | B to D | B to D | C to D | D to E | TOP3B |
| Sex | M | M | F | M | F | M | F | F | F | M | M | F | F | M |
| Age at last evaluation | 7 y | 11 y | 12 y | 12 y | 8 y | 1 y 3 mo | 1 y 8 mo | 27 y | 8 y | 3 y | 7 y | 14 y | 16 y | 6 y |
| Inheritance | De novo | De novo | ? | De novo | De novo | ? | De novo | ? | Maternal | ? | De novo | De novo | De novo | De novo |
| Dysmorphism | Mild | Atypic | + | Atypic | + | + | + | - | + | + | + | + | - | - |
| Microcephaly | - | + | - | - | - | Macrocephaly | + | - | - | - | + | - | - | - |
| Heart defects | VSD, ASD | VSD, CoA | VSD, ASD | ASD | - | - | TOF | - | TOF | TOF | VSD ASD | VSD | - | - |
| Palatal defects | CL, CP | VPI | - | CP | - | VPI | CP | - | - | - | - | - | VPI | CP |
| Development delay ± cognitive impairment | - | - | Mild | Moderate | Moderate | Severe | Moderate | - | - | - | Mild | - | - | - |
| Psychiatric/Behavior | - | ADHD | - | ADHD | ADHD | - | - | - | - | - | - | - | - | - |
| Hearing impairment | - | - | - | + | + | ± | ± | - | - | - | - | - | - | - |
| Growth delay | - | - | - | - | - | + | - | - | - | - | + | + | - | - |
| Hypotonia | - | - | - | + | - | - | - | - | - | - | - | - | - | - |
| Hypocalcemia | - | + | + | - | - | + | - | - | - | - | - | - | - | - |
| Seizures | - | + | + | - | - | + | - | - | ± | - | - | - | + | - |
| Immunodeficiency | - | - | - | - | + | + | + | - | - | - | - | - | - | - |
| Urogenital anomaly | - | - | - | RA, hypospadias | RA | HN | - | - | HN, RA | - | - | - | - | - |
| Other features | - | - | - | Auditory canal hypoplasia | Spastic tetraplegia | Cerebral atrophy, polydactyly | Spina bifida, polydactyly | - | - | - | CaL | CaL, hemangiomas | Progressive arthrosis | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavril, E.-C.; Popescu, R.; Nucă, I.; Ciobanu, C.-G.; Butnariu, L.I.; Rusu, C.; Pânzaru, M.-C. Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype–Phenotype Correlation. Genes 2022, 13, 2083. https://doi.org/10.3390/genes13112083
Gavril E-C, Popescu R, Nucă I, Ciobanu C-G, Butnariu LI, Rusu C, Pânzaru M-C. Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype–Phenotype Correlation. Genes. 2022; 13(11):2083. https://doi.org/10.3390/genes13112083
Chicago/Turabian StyleGavril, Eva-Cristiana, Roxana Popescu, Irina Nucă, Cristian-Gabriel Ciobanu, Lăcrămioara Ionela Butnariu, Cristina Rusu, and Monica-Cristina Pânzaru. 2022. "Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype–Phenotype Correlation" Genes 13, no. 11: 2083. https://doi.org/10.3390/genes13112083
APA StyleGavril, E. -C., Popescu, R., Nucă, I., Ciobanu, C. -G., Butnariu, L. I., Rusu, C., & Pânzaru, M. -C. (2022). Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype–Phenotype Correlation. Genes, 13(11), 2083. https://doi.org/10.3390/genes13112083