
The 22q11.2 Low Copy Repeats

Abstract
:1. Introduction
2. Instability of the LCR22s in the Human Genome
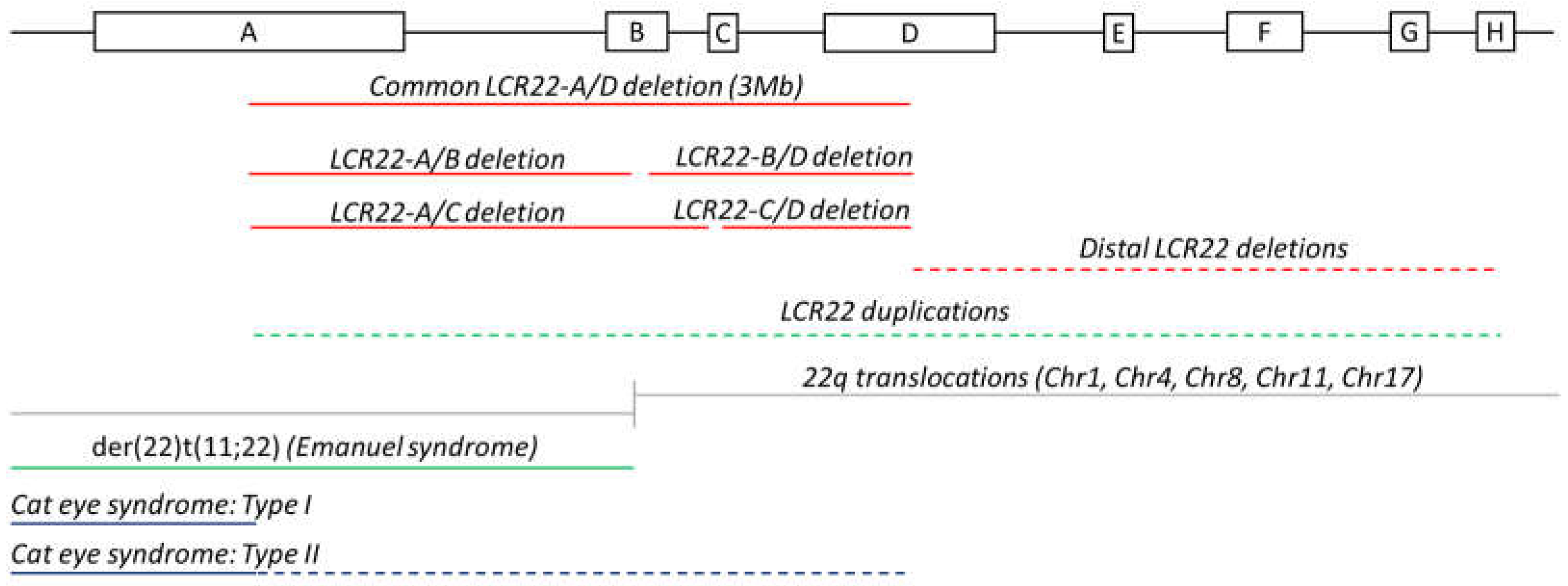
2.1. Deletions and Duplications
2.2. Translocations
2.3. Cat Eye Syndrome
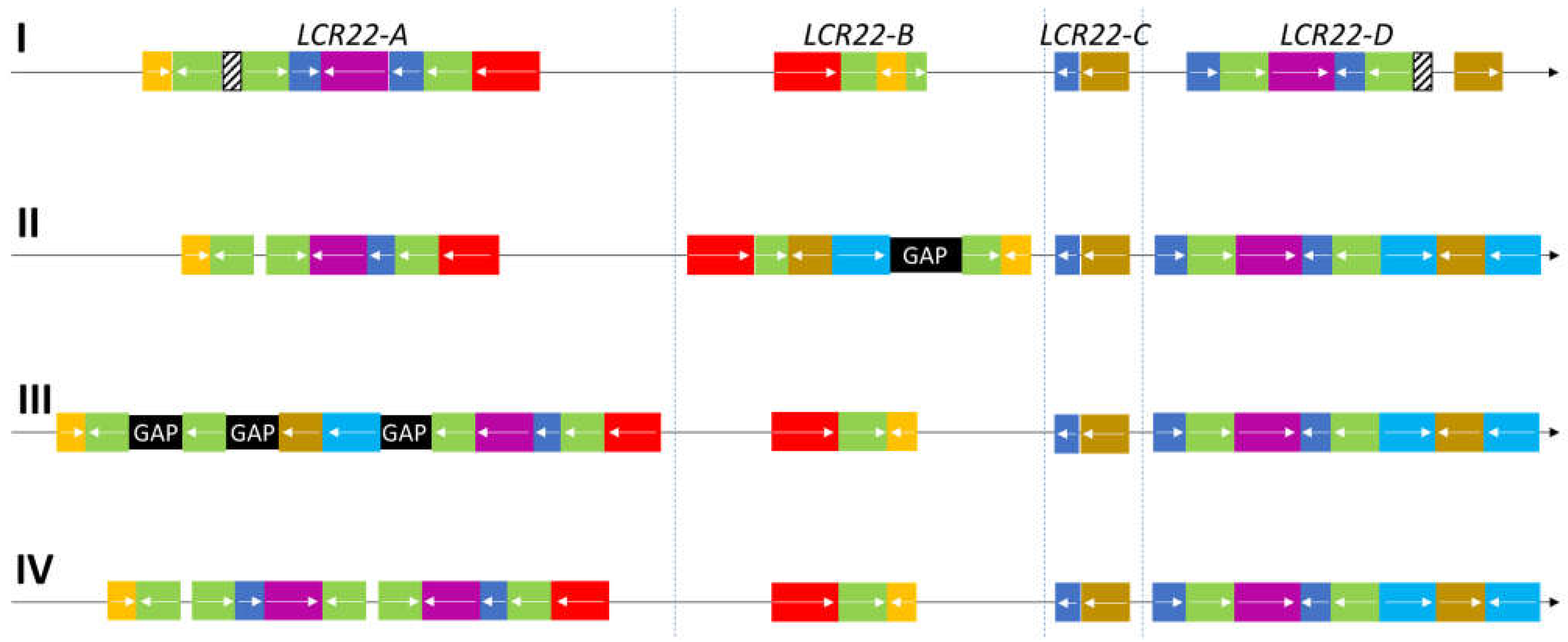
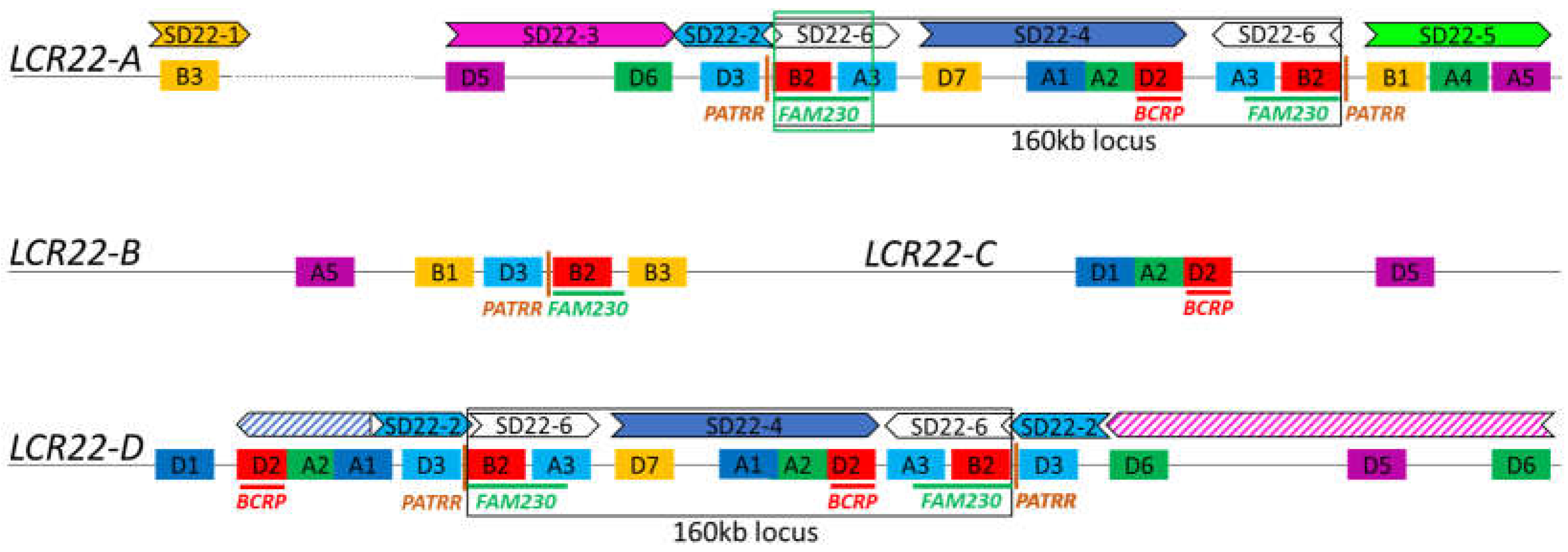
3. LCR22 Assembly and Representation in the Reference Genome
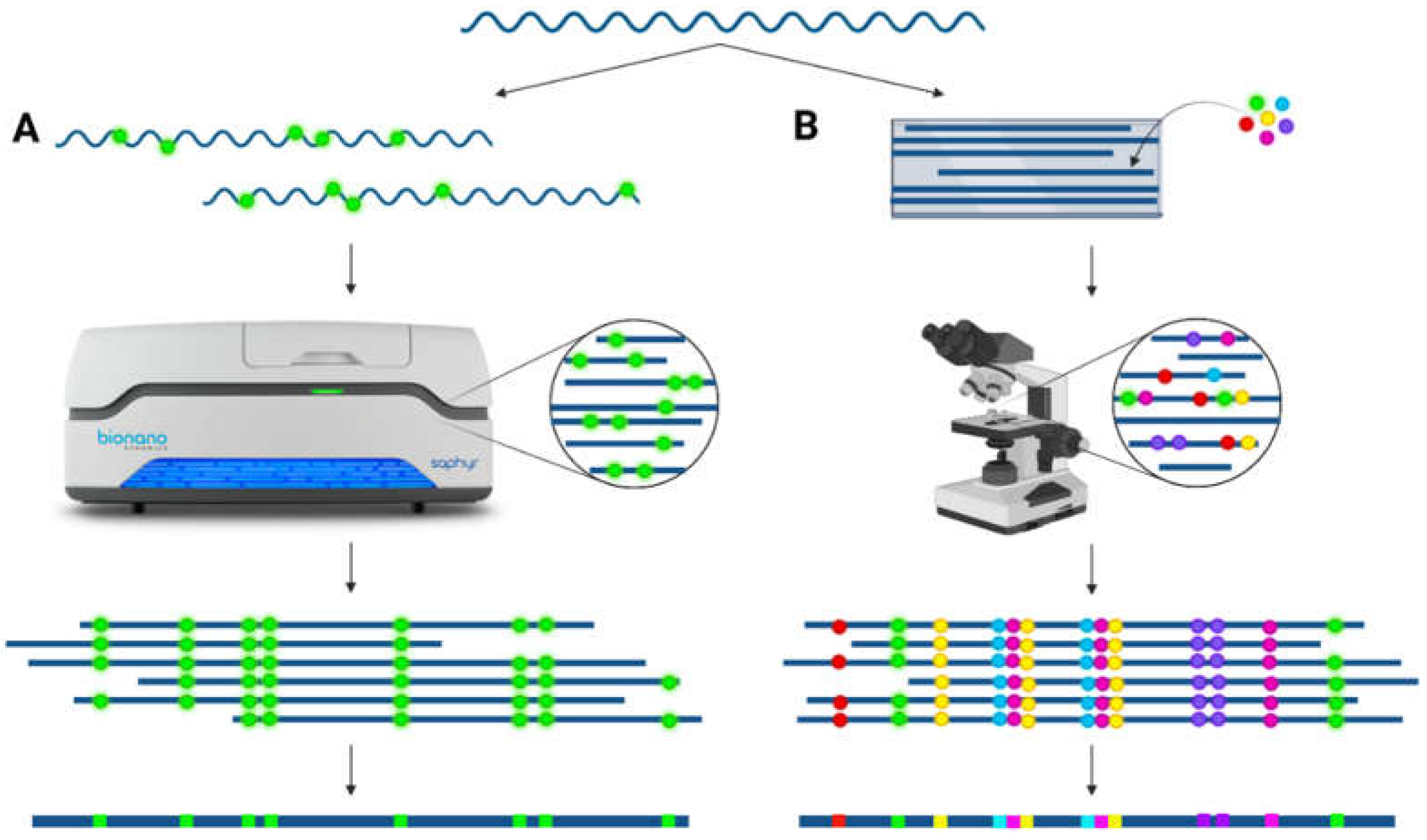
4. Entering the Era of Optical Mapping
5. The Hunt for the Rearrangement Sites
6. The Future: Ultralong Read Sequencing Approaches?
7. Remaining Questions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bailey, J.A.; Yavor, A.M.; Massa, H.F.; Trask, B.J.; Eichler, E.E. Segmental Duplications: Organization and Impact within the Current Human Genome Project Assembly. Genome Res. 2001, 11, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Gu, Z.; Clark, R.A.; Reinert, K.; Samonte, R.V.; Schwartz, S.; Adams, M.D.; Myers, E.W.; Li, P.W.; Eichler, E.E. Recent Segmental Duplications in the Human Genome. Science 2002, 297, 1003–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the Contemporary Live-Birth Prevalence of Recurrent 22q11.2 Deletions: A Cross-Sectional Analysis from Population-Based Newborn Screening. CMAJ Open 2021, 9, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Eichler, E.E. Human Adaptation and Evolution by Segmental Duplication. Curr. Opin. Genet. Dev. 2016, 41, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald-McGinn, D.; Sullivan, K.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.; Zackai, E.; Emanuel, B.; Vermeesch, J.; Morrow, B.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What Is New with 22q? An Update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. Part A 2018, 176, 2058–2069. [Google Scholar] [CrossRef]
- Burnside, R.D. 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet. Genome Res. 2015, 146, 89–99. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Ou, Z.; Shaw, C.A.; Belmont, J.W.; Patel, M.S.; Hummel, M.; Amato, S.; Tartaglia, N.; Berg, J.; Sutton, V.R.; et al. 22q11.2 Distal Deletion: A Recurrent Genomic Disorder Distinct from DiGeorge Syndrome and Velocardiofacial Syndrome. Am. J. Hum. Genet. 2008, 82, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Portnoï, M.F. Microduplication 22q11.2: A New Chromosomal Syndrome. Eur. J. Med. Genet. 2009, 52, 88–93. [Google Scholar] [CrossRef]
- Kato, T.; Kurahashi, H.; Emanuel, B.S. Chromosomal Translocations and Palindromic AT-Rich Repeats. Curr. Opin. Genet. Dev. 2012, 22, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, M.C.; Piotrowski, A.; Alexander, J.; Callens, T.; Fu, C.; Mikhail, F.M.; Claes, K.B.M.; Messiaen, L. Palindrome-Mediated and Replication-Dependent Pathogenic Structural Rearrangements within the NF1 Gene. Hum. Mutat. 2014, 35, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Budarf, M.L.; Celle, L.; Zackai, E.H.; Emanuel, B.S. Clustered 11q23 and 22q11 Breakpoints and 3:1 Meiotic Malsegregation in Multiple Unrelated t(11;22) Families. Am. J. Hum. Genet. 1999, 65, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenagui, R.; Bernicot, I.; Ranisavljevic, N.; Haquet, E.; Ferrieres-Hoa, A.; Pellestor, F.; Anahory, T. Inheritance of Imbalances in Recurrent Chromosomal Translocation t(11;22): Clarification by PGT-SR and Sperm-FISH Analysis. Reprod. Biomed. Online 2019, 39, 40–48. [Google Scholar] [CrossRef]
- Carter, M.T.; St. Pierre, S.A.; Zackai, E.H.; Emanuel, B.S.; Boycott, K.M. Phenotypic Delineation of Emanuel Syndrome (Supernumerary Derivative 22 Syndrome): Clinical Features of 63 Individuals. Am. J. Med. Genet. Part A 2009, 149A, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Emanuel, B.S. Molecular Mechanisms and Diagnosis of Chromosome 22q11.2 Rearrangements. Dev. Disabil. Res. Rev. 2008, 14, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia Chromosomal Breakpoints Are Clustered within a Limited Region, Bcr, on Chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations Involving 8q24 in Burkitt Lymphoma and Other Malignant Lymphomas: A Historical Review of Cytogenetics in the Light of Todays Knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Suva, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef]
- McDermid, H.E.; Morrow, B.E. Genomic Disorders on 22q11. Am. J. Hum. Genet. 2002, 70, 1077–1088. [Google Scholar] [CrossRef]
- Emanuel, B.S.; Shaikh, T.H. Segmental Duplications: An “expanding” Role in Genomic Instability and Disease. Nat. Rev. Genet. 2001, 2, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Shimizu, N.; Roe, B.A.; Chissoe, S.; Dunham, I.; Hunt, A.R.; Collins, J.E.; Bruskiewich, R.; Beare, D.M.; Clamp, M.; et al. The DNA Sequence of Human Chromosome 22. Nature 1999, 402, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.G.; McCann, O.T.; Collins, J.E.; Oliver, K.; Willey, D.; Gribble, S.M.; Yang, F.; McLaren, K.; Rogers, J.; Ning, Z.; et al. Finishing the Finished Human Chromosome 22 Sequence. Genome Biol. 2008, 9, R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-Specific Low Copy Repeats and the 22q11.2 Deletion Syndrome: Genomic Organization and Deletion Endpoint Analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the Complexity of the Human Genome Using Single-Molecule Sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar] [CrossRef]
- Mostovoy, Y.; Yilmaz, F.; Chow, S.K.; Chu, C.; Lin, C.; Geiger, E.A.; Meeks, N.J.L.; Chatfield, K.C.; Coughlin, C.R.; Surti, U.; et al. Genomic Regions Associated with Microdeletion/Microduplication Syndromes Exhibit Extreme Diversity of Structural Variation. Genetics 2021, 217, iyaa038. [Google Scholar] [CrossRef]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome Maps across 26 Human Populations Reveal Population-Specific Patterns of Structural Variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef] [Green Version]
- Louzada, S.; Komatsu, J.; Yang, F. Fluorescence In Situ Hybridization onto DNA Fibres Generated Using Molecular Combing. In Fluorescence In Situ Hybridization (FISH); Springer: Berlin/Heidelberg, Germany, 2017; pp. 275–293. [Google Scholar] [CrossRef]
- Algady, W.; Louzada, S.; Carpenter, D.; Brajer, P.; Färnert, A.; Rooth, I.; Ngasala, B.; Yang, F.; Shaw, M.A.; Hollox, E.J. The Malaria-Protective Human Glycophorin Structural Variant DUP4 Shows Somatic Mosaicism and Association with Hemoglobin Levels. Am. J. Hum. Genet. 2018, 103, 769–776. [Google Scholar] [CrossRef]
- Shi, W.; Massaia, A.; Louzada, S.; Handsaker, J.; Chow, W.; McCarthy, S.; Collins, J.; Hallast, P.; Howe, K.; Church, D.M.; et al. Birth, Expansion, and Death of VCY-Containing Palindromes on the Human Y Chromosome. Genome Biol. 2019, 20, 207. [Google Scholar] [CrossRef]
- Demaerel, W.; Mostovoy, Y.; Yilmaz, F.; Vervoort, L.; Pastor, S.; Hestand, M.S.; Swillen, A.; Vergaelen, E.; Geiger, A.; Coughlin, C.R.; et al. The 22q11 Low Copy Repeats Are Characterized by Unprecedented Size and Structural Variability. Genome Res. 2019, 29, 1389–1401. [Google Scholar] [CrossRef]
- Pastor, S.; Tran, O.; Jin, A.; Carrado, D.; Silva, B.A.; Uppuluri, L.; Abid, H.Z.; Young, E.; Crowley, T.B.; Bailey, A.G.; et al. Optical Mapping of the 22q11.2DS Region Reveals Complex Repeat Structures and Preferred Locations for Non-Allelic Homologous Recombination (NAHR). Sci. Rep. 2020, 10, 12235. [Google Scholar] [CrossRef]
- Vervoort, L.; Dierckxsens, N.; Pereboom, Z.; Capozzi, O.; Rocchi, M.; Shaikh, T.H.; Vermeesch, J.R. 22q11.2 Low Copy Repeats Expanded in the Human Lineage. Front. Genet. 2021, 12, 706641. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; O’Connor, R.J.; Pierpont, M.E.; McGrath, J.; Hacker, A.M.; Nimmakayalu, M.; Geiger, E.; Emanuel, B.S.; Saitta, S.C. Low Copy Repeats Mediate Distal Chromosome 22q11.2 Deletions: Sequence Analysis Predicts Breakpoint Mechanisms. Genome Res. 2007, 17, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Delio, M.; Haque, N.; Castellanos, R.; Hestand, M.S.; Vermeesch, J.R.; Morrow, B.E.; Zheng, D. Variant Discovery and Breakpoint Region Prediction for Studying the Human 22q11.2 Deletion Using BAC Clone and Whole Genome Sequencing Analysis. Hum. Mol. Genet. 2016, 25, 3754–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaujard, M.P.; Chantot, S.; Dubois, M.; Keren, B.; Carpentier, W.; Mabboux, P.; Whalen, S.; Vodovar, M.; Siffroi, J.P.; Portnoï, M.F. Atypical Deletion of 22q11.2: Detection Using the FISH TBX1 Probe and Molecular Characterization with High-Density SNP Arrays. Eur. J. Med. Genet. 2009, 52, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Amati, F.; Conti, E.; Novelli, A.; Bengala, M.; Digilio, M.C.; Marino, B.; Giannotti, A.; Gabrielli, O.; Novelli, G.; Dallapiccola, B. Atypical Deletions Suggest Five 22q11.2 Critical Regions Related to the DiGeorge/Velo-Cardio-Facial Syndrome. Eur. J. Hum. Genet. 1999, 7, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Stachon, A.C.; Squire, J.A.; Moldovan, L.; Bayani, J.; Meyn, S.; Chow, E.; Bassett, A.S. Molecular Characterization of Deletion Breakpoints in Adults with 22q11 Deletion Syndrome. Hum. Genet. 2007, 120, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.; Sirotkin, H.; Pandita, R.; Goldberg, R.; McKie, J.; Wadey, R.; Patanjali, S.R.; Weissman, S.M.; Anyane-Yeboa, K.; Warburton, D.; et al. Molecular Definition of 22q11 Deletions in 151 Velo-Cardio-Facial Syndrome Patients. Am. J. Hum. Genet. 1997, 61, 620–629. [Google Scholar] [CrossRef]
- Levy, A.; Demczuk, S.; Aurias, A.; Depétris, D.; Mattei, M.; Philip, N. Interstitial 22q11 Microdeletion Excluding the ADU Breakpoint in a Patient with DiGeorge Syndrome. Hum. Mol. Genet. 1995, 4, 2417–2419. [Google Scholar] [CrossRef] [Green Version]
- McQuade, L.; Christodoulou, J.; Budarf, M.L.; Sachdev, R.; Wilson, M.; Emanuel, B.; Colley, A. Patient with a 22q11.2 Deletion with No Overlap of the Minimal DiGeorge Syndrome Critical Region (MDGCR). Am. J. Hum. Genet. 1999, 3, 27–33. [Google Scholar] [CrossRef]
- Nogueira, S.I.; Hacker, A.M.; Bellucco, F.T.S.; Christofolini, D.M.; Kulikowski, L.D.; Cernach, M.C.S.P.; Emanuel, B.S.; Melaragno, M.I. Atypical 22q11.2 Deletion in a Patient with DGS/VCFS Spectrum. Eur. J. Med. Genet. 2008, 51, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, H.; McKeown, C.; Gould, C.; Morrow, B.; Scambler, P. Detection of an Atypical 22q11 Deletion That Has No Overlap with the DiGeorge Syndrome Critical Region. Am. J. Hum. Genet 1997, 60, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.K.; Zhang, Y.; Siu, V.M.; Fan, Y.S.; O’Reilly, R.L.; Rao, J.; Singh, S.M. Breakpoint Associated with a Novel 2.3 Mb Deletion in the VCFS Region of 22q11 and the Role of Alu (SINE) in Recurring Microdeletions. BMC Med. Genet. 2006, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gur, R.E.; Bassett, A.S.; McDonald-Mcginn, D.M.; Bearden, C.E.; Chow, E.; Emanuel, B.S.; Owen, M.; Swillen, A.; den Bree, M.V.; Vermeesch, J.; et al. A Neurogenetic Model for the Study of Schizophrenia Spectrum Disorders: The International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol. Psychiatry 2017, 22, 1664–1672. [Google Scholar] [CrossRef]
- Vervoort, L.; Demaerel, W.; Rengifo, L.Y.; Odrzywolski, A.; Vergaelen, E.; Hestand, M.S.; Breckpot, J.; Devriendt, K.; Swillen, A.; McDonald-McGinn, D.M.; et al. Atypical Chromosome 22q11.2 Deletions Are Complex Rearrangements and Have Different Mechanistic Origins. Hum. Mol. Genet. 2019, 28, 3724–3733. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A Graphical Viewer for Oxford Nanopore Bulk FAST5 Files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Beyter, D.; Ingimundardottir, H.; Oddsson, A.; Eggertsson, H.P.; Bjornsson, E.; Jonsson, H.; Atlason, B.A.; Kristmundsdottir, S.; Mehringer, S.; Hardarson, M.T.; et al. Long-Read Sequencing of 3,622 Icelanders Provides Insight into the Role of Structural Variants in Human Diseases and Other Traits. Nat. Genet. 2021, 53, 779–786. [Google Scholar] [CrossRef]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.P.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.D.W.; et al. Long-Read Sequence Assembly of the Gorilla Genome. Science 2016, 352, aae0344. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Catacchio, C.R.; Hillier, L.D.W.; Porubsky, D.; Li, R.; Sulovari, A.; Fernandes, J.D.; Montinaro, F.; Gordon, D.S.; Storer, J.M.; et al. A High-Quality Bonobo Genome Refines the Analysis of Hominid Evolution. Nature 2021, 594, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Antonacci-Fulton, L.; Howe, K.; Lawson, H.A.; Lucas, J.K.; Phillippy, A.M.; Popejoy, A.B.; Asri, M.; Carson, C.; Chaisson, M.J.P.; et al. The Human Pangenome Project: A Global Resource to Map Genomic Diversity. Nature 2022, 604, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Zhu, X.; Purmann, C.; Haney, M.S.; Ward, T.; Khechaduri, A.; Yao, J.; Weissman, S.M.; Urban, A.E. Local and Global Chromatin Interactions Are Altered by Large Genomic Deletions Associated with Human Brain Development. Nat. Commun. 2018, 9, 5356. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GRCh37/hg19 | GRCh38/hg38 | T2T-CHM13 | |
|---|---|---|---|
| LCR22-A | chr22:18,639,043-19,022,986 | chr22:18,156,276-19,035,473 | CP068256.2:18,828,186-19,410,796 |
| LCR22-B | chr22:20,128,537-20,731,921 | chr22:20,141,014-20,377,631 | CP068256.2:20,520,047-20,781,953 |
| LCR22-C | chr22:21,021,564-21,092,560 | chr22:20,667,276-20,738,272 | CP068256.2:21,075,991-21,146,982 |
| LCR22-D | chr22:21,363,668-21,916,380 | chr22:21,009,379-21,562,091 | CP068256.2:21,418,153-21,975,566 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vervoort, L.; Vermeesch, J.R. The 22q11.2 Low Copy Repeats. Genes 2022, 13, 2101. https://doi.org/10.3390/genes13112101
Vervoort L, Vermeesch JR. The 22q11.2 Low Copy Repeats. Genes. 2022; 13(11):2101. https://doi.org/10.3390/genes13112101
Chicago/Turabian StyleVervoort, Lisanne, and Joris Robert Vermeesch. 2022. "The 22q11.2 Low Copy Repeats" Genes 13, no. 11: 2101. https://doi.org/10.3390/genes13112101
APA StyleVervoort, L., & Vermeesch, J. R. (2022). The 22q11.2 Low Copy Repeats. Genes, 13(11), 2101. https://doi.org/10.3390/genes13112101