
Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Classification of Ectodermal Dysplasias and Their Correct Diagnosis
3. Phenotype
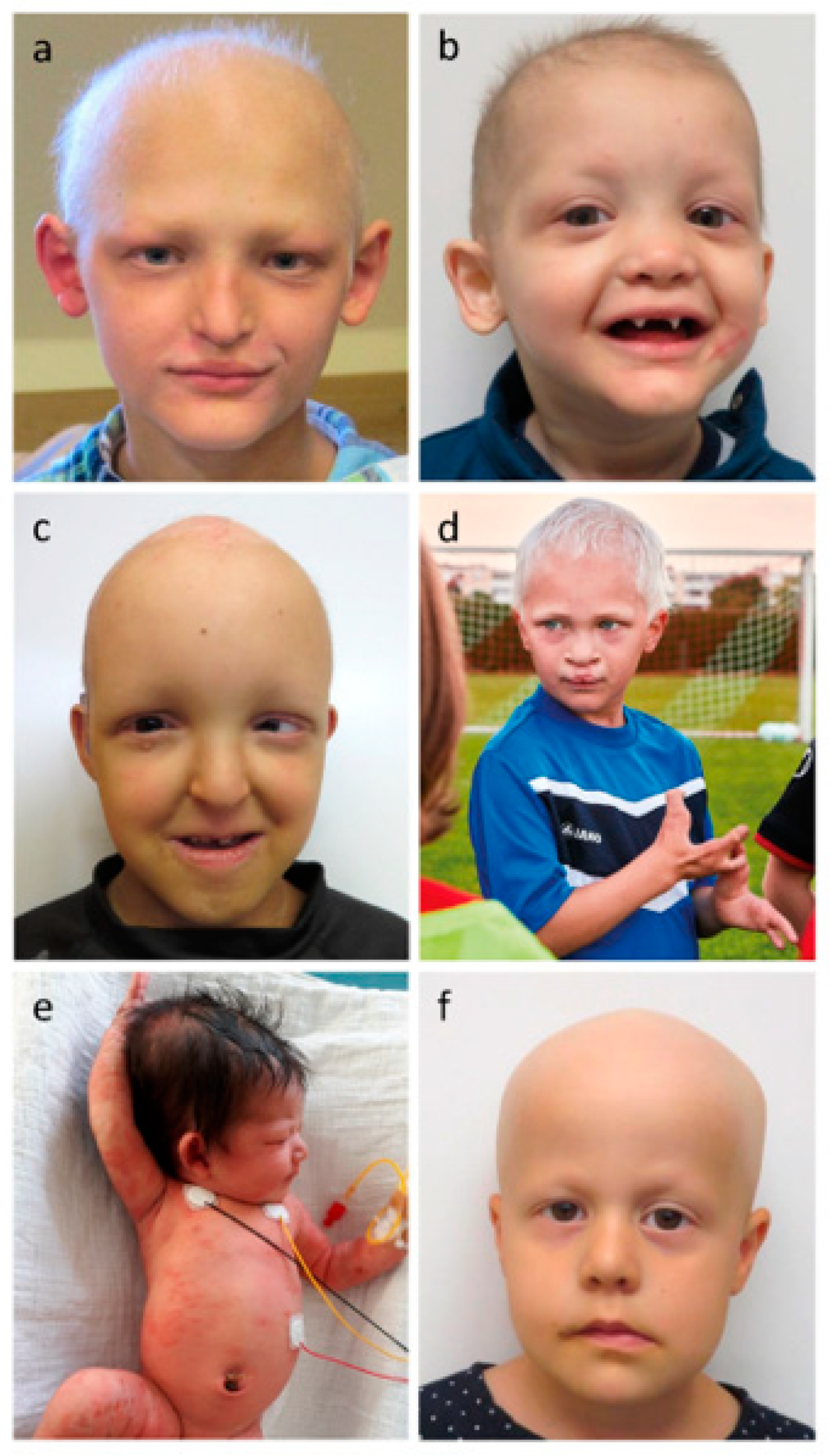
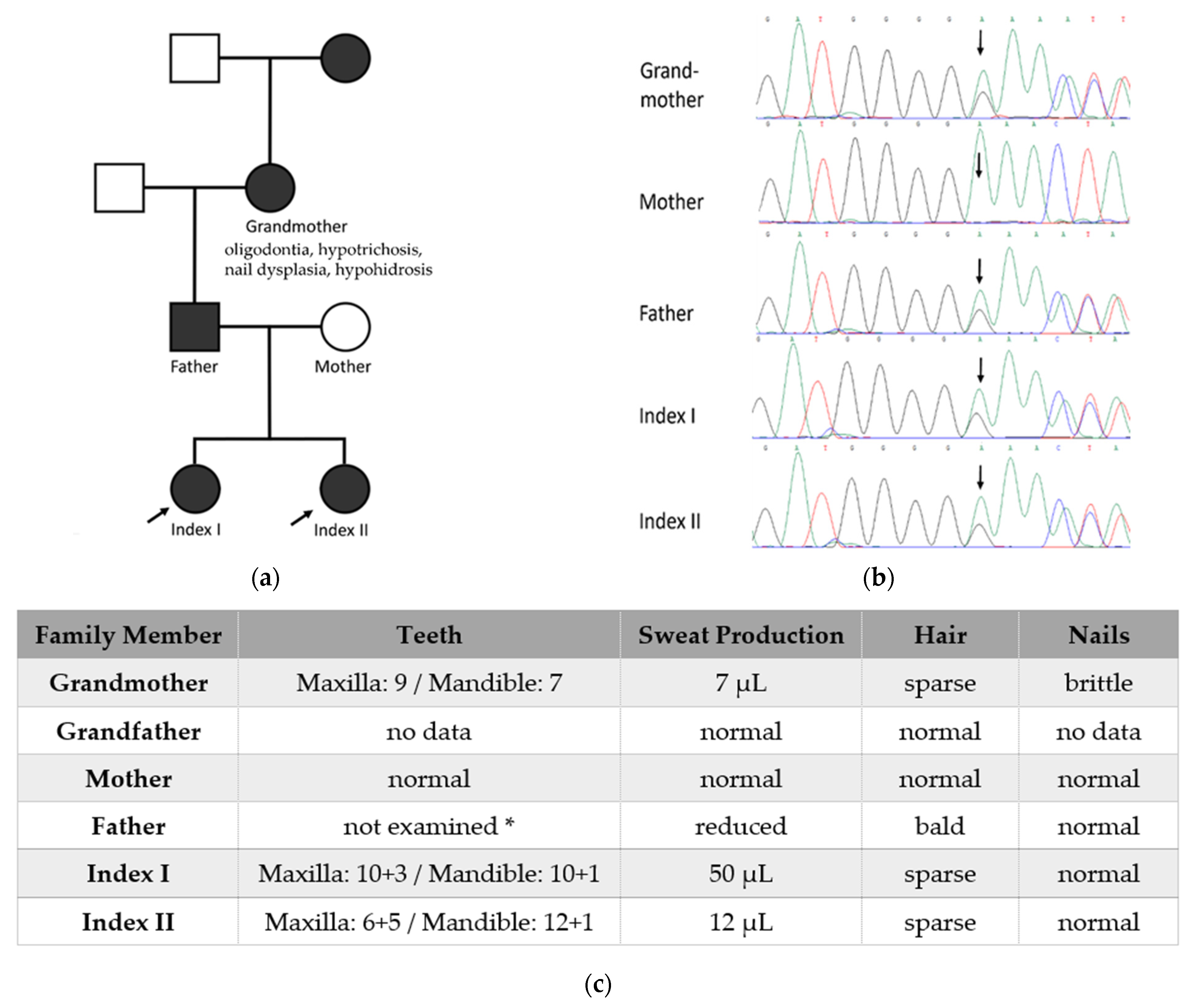
3.1. Glandular Phenotypes
3.2. Hair Phenotypes
3.3. Dental Phenotypes
3.4. Nail Phenotypes
3.5. Other Manifestations
4. Molecular Signaling Pathways
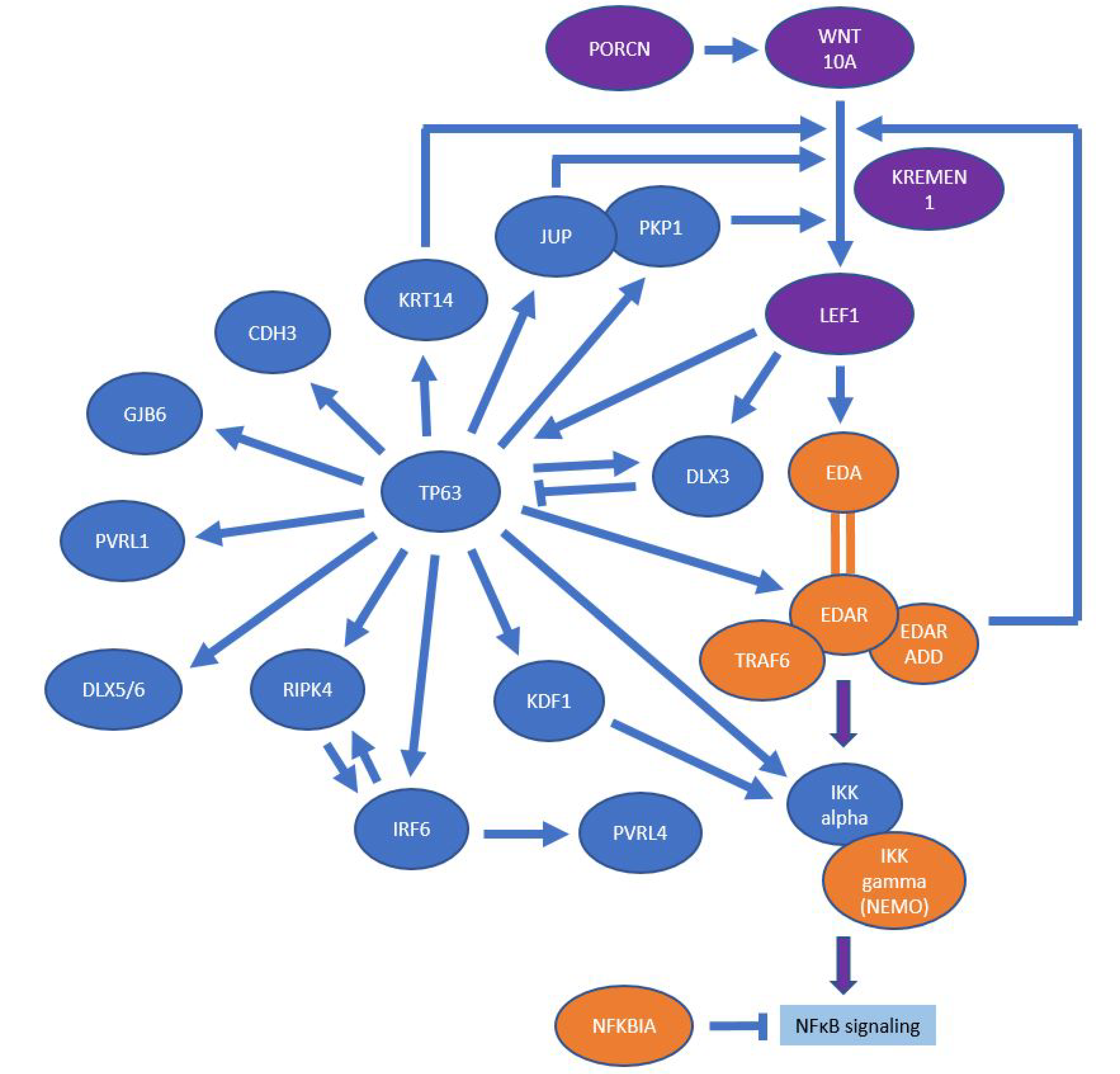
4.1. EDA/NF-κB Pathway
4.2. Wnt/β-Catenin Pathway
4.3. p63 Signaling
4.4. Keratinization and Structure-Giving Proteins
5. Selection of Diseases to Be Classified
5.1. Exclusions
5.2. New Inclusions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freire-Maia, N. Nosologic groups. An overview. Hum. Hered. 1977, 27, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Pagnan, N.A.B.; Visinoni, Á.F. Update on ectodermal dysplasias clinical classification. Am. J. Med. Genet. A 2014, 164A, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. A 2019, 179, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Itin, P.H. Etiology and pathogenesis of ectodermal dysplasias. Am. J. Med. Genet. A 2014, 164A, 2472–2477. [Google Scholar] [CrossRef]
- Nguyen-Nielsen, M.; Skovbo, S.; Svaneby, D.; Pedersen, L.; Fryzek, J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur. J. Med. Genet. 2013, 56, 236–242. [Google Scholar] [CrossRef]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.-C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Körber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal correction of X-linked hypohidrotic ectodermal dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef]
- Murdock, S.; Lee, J.Y.; Guckes, A.; Wright, J.T. A costs analysis of dental treatment for ectodermal dysplasia. J. Am. Dent. Assoc. 2005, 136, 1273–1276. [Google Scholar] [CrossRef]
- Asano, N.; Yasuno, S.; Hayashi, R.; Shimomura, Y. Characterization of EDARADD gene mutations responsible for hypohidrotic ectodermal dysplasia. J. Dermatol. 2021, 48, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, H.; Farooq, M.; Fujimoto, A.; Ito, M.; Shimomura, Y. Functional studies for the TRAF6 mutation associated with hypohidrotic ectodermal dysplasia. Br. J. Dermatol. 2013, 168, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Capri, Y.; Rachid, M.; Dupont, C.; Vermeesch, J.R.; Devriendt, K.; Verloes, A.; Tabet, A.-C.; Bailleul-Forestier, I. LEF1 haploinsufficiency causes ectodermal dysplasia. Clin. Genet. 2020, 97, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Dufour, W.; Alawbathani, S.; Jourdain, A.-S.; Asif, M.; Baujat, G.; Becker, C.; Budde, B.; Gallacher, L.; Georgomanolis, T.; Ghoumid, J.; et al. Monoallelic and biallelic variants in LEF1 are associated with a new syndrome combining ectodermal dysplasia and limb malformations caused by altered WNT signaling. Genet. Med. 2022, 24, 1708–1721. [Google Scholar] [CrossRef]
- Yu, M.; Fan, Z.; Wong, S.W.; Sun, K.; Zhang, L.; Liu, H.; Feng, H.; Liu, Y.; Han, D. Lrp6 dynamic expression in tooth development and mutations in oligodontia. J. Dent. Res. 2021, 100, 415–422. [Google Scholar] [CrossRef]
- Massey, H.; House, J.; Tipton, M. Thermoregulation in ectodermal dysplasia: A case series. Int. J. Environ. Res. Public Health 2019, 16, 4514. [Google Scholar] [CrossRef] [Green Version]
- Blüschke, G.; Nüsken, K.-D.; Schneider, H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum. Dev. 2010, 86, 397–399. [Google Scholar] [CrossRef]
- Minor, V. Ein neues Verfahren zu der klinischen Untersuchung der Schweißabsonderung. Deutsch. Z. Nervenheilkd. 1928, 101, 302–308. [Google Scholar] [CrossRef]
- Schneider, H.; Hammersen, J.; Preisler-Adams, S.; Huttner, K.; Rascher, W.; Bohring, A. Sweating ability and genotype in individuals with X-linked hypohidrotic ectodermal dysplasia. J. Med. Genet. 2011, 48, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Kaercher, T.; Dietz, J.; Jacobi, C.; Berz, R.; Schneider, H. Diagnosis of X-linked hypohidrotic ectodermal dysplasia by meibography and infrared thermography of the eye. Curr. Eye Res. 2015, 40, 884–890. [Google Scholar] [CrossRef]
- Keklikci, U.; Yavuz, I.; Tunik, S.; Ulku, Z.B.; Akdeniz, S. Ophthalmic manifestations in patients with ectodermal dysplasia syndromes. Adv. Clin. Exp. Med. 2014, 23, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semmler, M.; Kniesburges, S.; Pelka, F.; Ensthaler, M.; Wendler, O.; Schützenberger, A. Influence of reduced saliva production on phonation in patients with ectodermal dysplasia. J. Voice 2021, in press. [Google Scholar] [CrossRef]
- Nordgarden, H.; Storhaug, K.; Lyngstadaas, S.P.; Jensen, J.L. Salivary gland function in persons with ectodermal dysplasias. Eur. J. Oral Sci. 2003, 111, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wahlbuhl-Becker, M.; Faschingbauer, F.; Beckmann, M.W.; Schneider, H. Hypohidrotic ectodermal dysplasia: Breastfeeding complications due to impaired breast development. Geburtshilfe Frauenheilkd. 2017, 77, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, R.; Shimomura, Y. Update of recent findings in genetic hair disorders. J. Dermatol. 2022, 49, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Peña-Romero, A.G.; Sáez-de-Ocariz, M.; Toussaint-Caire, S.; Morán-Villaseñor, E.; Orozco-Covarrubias, L.; Durán-McKinster, C. Clinical, trichoscopy, and light microscopic findings in hypohidrotic ectodermal dysplasia: Report of 21 patients and a review of the literature. Pediatr. Dermatol. 2021, 38, 442–448. [Google Scholar] [CrossRef]
- Hirano-Ali, S.A.; Reed, A.M.; Rowan, B.J.; Sorrells, T.; Williams, J.V.; Pariser, D.M.; Hood, A.F.; Salkey, K. Scanning electron microscopic hair shaft analysis in ectodermal dysplasia syndromes. Pediatr. Dermatol. 2015, 32, 836–844. [Google Scholar] [CrossRef]
- Bergendal, B. Orodental manifestations in ectodermal dysplasia-a review. Am. J. Med. Genet. A 2014, 164A, 2465–2471. [Google Scholar] [CrossRef]
- Price, J.A.; Wright, J.T.; Walker, S.J.; Crawford, P.J.; Aldred, M.J.; Hart, T.C. Tricho-dento-osseous syndrome and amelogenesis imperfecta with taurodontism are genetically distinct conditions. Clin. Genet. 1999, 56, 35–40. [Google Scholar] [CrossRef]
- García-Martín, P.; Hernández-Martín, A.; Torrelo, A. Ectodermal dysplasias: A clinical and molecular review. Actas Dermo-Sifiliogr. (English Edition) 2013, 104, 451–470. [Google Scholar] [CrossRef]
- Visinoni, A.F.; Lisboa-Costa, T.; Pagnan, N.A.B.; Chautard-Freire-Maia, E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. A 2009, 149A, 1980–2002. [Google Scholar] [CrossRef]
- Shimomura, Y.; Wajid, M.; Shapiro, L.; Christiano, A.M. P-cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development 2008, 135, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements. J. Hum. Genet. 2016, 61, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Doolan, B.J.; Onoufriadis, A.; Kantaputra, P.; McGrath, J.A. WNT10A, dermatology and dentistry. Br. J. Dermatol. 2021, 185, 1105–1111. [Google Scholar] [CrossRef]
- Ezer, S.; Bayés, M.; Elomaa, O.; Schlessinger, D.; Kere, J. Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum. Mol. Genet. 1999, 8, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morlon, A.; Munnich, A.; Smahi, A. TAB2, TRAF6 and TAK1 are involved in NF-kappaB activation induced by the TNF-receptor, Edar and its adaptator Edaradd. Hum. Mol. Genet. 2005, 14, 3751–3757. [Google Scholar] [CrossRef] [Green Version]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Verhelst, K.; Gardam, S.; Borghi, A.; Kreike, M.; Carpentier, I.; Beyaert, R. XEDAR activates the non-canonical NF-κB pathway. Biochem. Biophys. Res. Commun. 2015, 465, 275–280. [Google Scholar] [CrossRef]
- Yu, M.; Wong, S.-W.; Han, D.; Cai, T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019, 25, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Koster, M.I. p63 in skin development and ectodermal dysplasias. J. Investig. Dermatol. 2010, 130, 2352–2358. [Google Scholar] [CrossRef]
- Mitchell, K.; O’Sullivan, J.; Missero, C.; Blair, E.; Richardson, R.; Anderson, B.; Antonini, D.; Murray, J.C.; Shanske, A.L.; Schutte, B.C.; et al. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am. J. Hum. Genet. 2012, 90, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Tanis, S.E.J.; Smits, J.P.H.; Kouwenhoven, E.N.; Oti, M.; van den Bogaard, E.H.; Logie, C.; Stunnenberg, H.G.; van Bokhoven, H.; Mulder, K.W.; et al. Mutant p63 affects epidermal cell identity through rewiring the enhancer landscape. Cell Rep. 2018, 25, 3490–3503.e4. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.; Thompson, B.; Fisk, J.N.; Nebert, D.W.; Bruford, E.A.; Vasiliou, V.; Bunick, C.G. Update of the keratin gene family: Evolution, tissue-specific expression patterns, and relevance to clinical disorders. Hum. Genom. 2022, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Petrof, G.; Mellerio, J.E.; McGrath, J.A. Desmosomal genodermatoses. Br. J. Dermatol. 2012, 166, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Guo, K.; Li, J.; Liu, Y.; Zeng, W.H.; Geng, S.M. Novel mutations in GJB6 and GJB2 in Clouston syndrome. Clin. Exp. Dermatol. 2015, 40, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Ellingson, R.J. Major hereditary ectodermal dysplasia. J. Pediatr. 1951, 38, 191–198. [Google Scholar] [CrossRef]
- Peled, A.; Sarig, O.; Samuelov, L.; Bertolini, M.; Ziv, L.; Weissglas-Volkov, D.; Eskin-Schwartz, M.; Adase, C.A.; Malchin, N.; Bochner, R.; et al. Mutations in TSPEAR, encoding a regulator of Notch signaling, affect tooth and hair follicle morphogenesis. PLoS Genet. 2016, 12, e1006369. [Google Scholar] [CrossRef] [Green Version]
- Bowles, B.; Ferrer, A.; Nishimura, C.J.; Pinto, E.; Vairo, F.; Rey, T.; Leheup, B.; Sullivan, J.; Schoch, K.; Stong, N.; et al. TSPEAR variants are primarily associated with ectodermal dysplasia and tooth agenesis but not hearing loss: A novel cohort study. Am. J. Med. Genet. A 2021, 185, 2417–2433. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, Q.; Shi, L.; Lee, M.; Giehl, K.A.; Tang, Z.; Wang, H.; Zhang, J.; Yin, J.; Wu, L.; et al. Loss-of-function mutations in HOXC13 cause pure hair and nail ectodermal dysplasia. Am. J. Hum. Genet. 2012, 91, 906–911. [Google Scholar] [CrossRef] [Green Version]
- van den Bogaard, E.H.J.; van Geel, M.; van Vlijmen-Willems, I.M.J.J.; Jansen, P.A.M.; Peppelman, M.; van Erp, P.E.J.; Atalay, S.; Venselaar, H.; Simon, M.E.H.; Joosten, M.; et al. Deficiency of the human cysteine protease inhibitor cystatin M/E causes hypotrichosis and dry skin. Genet. Med. 2019, 21, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Wehrli, M.; Dougan, S.T.; Caldwell, K.; O’Keefe, L.; Schwartz, S.; Vaizel-Ohayon, D.; Schejter, E.; Tomlinson, A.; DiNardo, S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature 2000, 407, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A mechanism for Wnt coreceptor activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. A finger on the pulse of Wnt receptor signaling. Cell Res. 2012, 22, 1410–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; van Rooij, I.A.L.M.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Chae, W.; Kim, Y.J.; Kim, J.-W. Novel LRP6 mutations causing non-syndromic oligodontia. J. Pers. Med. 2022, 12, 1401. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Affected | Disease(s)/Syndrome(s)—Previously Suggested Nomenclature | OMIM No. | Hypo- | Hair | Nail | Glandular | Additional | |
|---|---|---|---|---|---|---|---|---|
| Gene(s) | dontia | Phenotypic Features | Symptoms | |||||
| EDA-NFκB pathway | ||||||||
| EDA | Ectodermal dysplasia 1, hypohidrotic, X-linked (Christ–Siemens–Touraine syndrome, XLHED) | 305100 | ● | ● | ● | |||
| EDAR | Ectodermal dysplasia 10A and 10B, AD and AR (ECTD10A, B) | 129490, 224900 | ● | ● | ● | ● | ||
| EDARADD | Ectodermal dysplasia 11A and 11B, AD and AR (ECTD11A, B) | 614940, 614941 | ● | ● | ● | ● | ||
| IKBKG | Incontinentia pigmenti (IP) | 308300 | ● | ● | ● | |||
| IKBKG | Ectodermal dysplasia and immunodeficiency 1, AD and AR (EDAID1) | 300291, 300301 | ● | ● | ● | |||
| CHUK | Cocoon syndrome | 613630 | ● | ● | ● | ● | ||
| NFKBIA | Ectodermal dysplasia and immunodeficiency 2 (EDAID2) | 612132 | ● | ● | ● | |||
| PRKD1 | Congenital heart defects and ectodermal dysplasia (CHDED) | 617364 | ● | ● | ● | ● | ||
| TRAF6 | Hidrotic form of ectodermal dysplasia, not yet OMIM-listed 1 | 602355 | ● | ● | ● | |||
| WNT pathway | PORCN | Focal dermal hypoplasia (FDH) or Goltz–Gorlin syndrome | 305600 | ● | ● | ● | ● | ● |
| TWIST2 | Focal facial dermal dysplasia 3/Ablepharon-macrostomia syndrome (AMS) | 136500, 200110 | ● | ● | ● | |||
| WNT10A | Odonto–onycho–dermal dysplasia (OODD)/Schöpf–Schulz–Passarge syndrome (SSPS) | 257980, 224750 | ● | ● | ● | ● | ||
| KREMEN1 | Ectodermal dysplasia 13, hair/tooth type (ECTD13) | 617392 | ● | ● | ||||
| TBX3 | Ulnar-mammary syndrome (UMS) | 181450 | ● | ● | ● | ● | ||
| LEF1 | Ectodermal dysplasia with or without hypohidrosis, not yet OMIM-listed 2 | 153245 | ● | ● | ● | ● | ||
| LRP6 | Ectodermal dysplasia with or without hypohidrosis, not yet OMIM-listed 3 | (603507) * | ● | ● | ● | ● | ||
| MSX1 | Ectodermal dysplasia 3, Witkop type (ECTD3 or Witkop syndrome) | 189500 | ● | ● | ||||
| p63 pathway | TP63 | Acro–dermato–ungual–lacrimal–tooth syndrome (ADULT) | 103285 | ● | ● | ● | ● | |
| TP63 | Ectrodactyly ectodermal dysplasia–cleft lip/palate syndrome (EEC) | 604292 | ● | ● | ● | ● | ● | |
| TP63 | Limb–mammary syndrome (LMS) | 603543 | ● | ● | ● | ● | ||
| TP63 | Ankyloblepharon–ectodermal defects–cleft lip/palate (AEC/Rapp–Hodgkin syndrome) | 106260, 129400 | ● | ● | ● | ● | ● | |
| CDH3 | Ectodermal dysplasia, ectrodactyly, macular dystrophy syndrome (EEMS) | 225280 | ● | ● | ● | |||
| KDF1 | Ectodermal dysplasia 12, hypohidrotic/hair/tooth/nail type (ECTD12) | 617337 | ● | ● | ● | ● | ||
| DLX3 | Tricho–dento–osseous syndrome (TDO) | 190320 | ● | ● | ||||
| RIPK4 | Curly hair–ankyloblepharon–nail dysplasia syndrome (CHANDS);complex lethal subtype known as Bartsocas–Papas syndrome 1 (BPS1) | 214350, 263650 | ● | ● | ● | |||
| Structure group | PKP1 | Ectodermal dysplasia–skin fragility syndrome (EDSFS) | 604536 | ● | ● | ● | ● | |
| GRHL2 | Ectodermal dysplasia–short stature syndrome (ECTDS) | 616029 | ● | ● | ● | |||
| PVRL1 | Cleft lip/palate–ectodermal dysplasia syndrome (CLPED1) | 225060 | ● | ● | ● | ● | ||
| PVRL4 | Ectodermal dysplasia–syndactyly syndrome 1 (EDSS1) | 613573 | ● | ● | ● | ● | ||
| KRT74 | Ectodermal dysplasia 7, hair/nail type (ECTD7) | 614929 | ● | ● | ||||
| KRT85 | Ectodermal dysplasia 4, hair/nail type (ECTD4) | 602032 | ● | ● | ||||
| GJB2 | Keratitis–ichthyosis–deafness syndrome, AD (KID) | 148210 | ● | ● | ● | |||
| GJA1 | Oculo–dento–digital dysplasia (ODDD, ODDR) | 164200, 257850 | ● | ● | ● | ● | ● | |
| GJB6 | Clouston syndrome or ectodermal dysplasia 2, Clouston type (ECTD2) | 129500 | ● | ● | ||||
| Others | TSPEAR | Ectodermal dysplasia 14, hair/tooth type with or without hypohidrosis (ECTD14) | 618180 | ● | ● | ● | ● | |
| HOXC13 | Ectodermal dysplasia 9, hair/nail type (ECTD9) | 614931 | ● | ● | ||||
| CST6 | Ectodermal dysplasia 1, hypohidrotic/hair type (ECTD15) | 618535 | ● | ● | ||||
| AP1B1 | Keratitis–ichthyosis–deafness syndrome, AR (KIDAR) | 242150 | ● | ● | ● | ● | ● | |
| TRPS1 | Trichorhinophalangeal syndrome, type I (TRPS1) or type III (TRPS3) | 190350, 190351 | ● | ● | ● | ● | ||
| TRPS1 + EXT1 | Trichorhinophalangeal syndrome, type II (TRPS2) | 150230 | ● | ● | ● | ● | ||
| Affected |
Syndrome(s) with a Partial Ectodermal Dysplasia-Like Phenotype but Classified Elsewhere | OMIM No. | Hypo- | Hair | Nail | Glandular | Additional |
|---|---|---|---|---|---|---|---|
| Gene(s) | dontia | Phenotypic Features | Symptoms | ||||
| CDH1, CTNND1 | Blepharocheilodontic syndrome 1/Blepharocheilodontic syndrome 2 (BCDS1/BCDS2) | 119580, 617681 | ● | ● | ● | ||
| IFT43, -52, -122, -140 WDR19, -35 | Cranioectodermal dysplasia, types 1–4 (CED) or Sensenbrenner syndrome | 218330 | ● | ● | ● | ● | |
| EVC, EVC2 | Ellis–van Creveld syndrome (EVC) | 225500 | ● | ● | ● | ● | |
| EVC, EVC2 | Weyers acrofacial dysostosis (WAD) | 193530 | ● | ● | ● | ||
| KCTD1 | Scalp–ear–nipple syndrome (SENS) | 181270 | ● | ● | ● | ● | ● |
| SOX9–CNJ2 * | Cooks syndrome | 106995 | ● | ● | ● | ||
| ANTXR1 | Growth retardation, alopecia, pseudoanodontia, and optic atrophy syndrome (GAPO) | 230740 | ● | ● | ● | ● | |
| SMARCAD1 | Huriez syndrome/Basan syndrome | 181600, 129200 | ● | ● | |||
| DSP | Carvajal syndrome (DCWHK) | 605676 | ● | ● | ● | ● | |
| KRT14 | Dermatopathia pigmentosa reticularis (DPR)/Naegeli syndrome (NFJS) | 125595, 161000 | ● | ● | ● | ● | |
| KRT16, -17 | Pachyonychia congenita 1/Pachyonychia congenita 2 (PC1/PC2) | 167200, 167210 | ● | ● | ● | ● | |
| ARID1A, -1B SMARCA4, -B1, -E1 | Coffin–Siris syndrome (CSS) | 135900 | ● | ● | ● | ● | |
| ATP6V1B2 | Deafness, congenital, and onychodystrophy, AD (DDOD) | 124480 | ● | ● | ● | ||
| TBC1D24 | Deafness, onycho- and osteodystrophy, mental retardation, and seizures syndrome | 220500 | ● | ● | ● | ||
| SLC25A24 | Gorlin–Chaudhry–Moss syndrome (FPS) | 612289 | ● | ● | ● | ● | |
| PEX1, PEX6 | Heimler syndrome 1 (HMLR1)/Heimler syndrome 2 (HMLR2) | 234580, 616617 | ● | ● | ● | ||
| UBR1 | Johanson–Blizzard syndrome (JBS) | 243800 | ● | ● | ● | ||
| FGFR3, -2 FGF10 | Lacrimo–auriculo–dento–digital syndrome (LADD) | 149730 | ● | ● | ● | ||
| SREBF1 | Mucoepithelial dysplasia, hereditary (HMD) | 158310 | ● | ● | |||
| HEPHL1 | Pili torti and developmental delay (HJDD) | 261990 | ● | ● | ● | ||
| KRT81, -83, -86, DSG4 | Monilethrix (MNLIX) | 158000 | ● | ● | |||
| RODGI | Kohlschütter–Tönz syndrome | 226750 | ● | ● | ● | ||
| INSR | Pineal hyperplasia, insulin-resistant diabetes mellitus, and somatic abnormalities | 262190 | ● | ● | ● | ● | |
| CTSK | Pycnodysostosis (PKND) | 265800 | ● | ● | ● | ||
| SETBP1 | Schinzel–Giedion midface retraction syndrome (SGS) | 269150 | ● | ● | ● | ● | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peschel, N.; Wright, J.T.; Koster, M.I.; Clarke, A.J.; Tadini, G.; Fete, M.; Hadj-Rabia, S.; Sybert, V.P.; Norderyd, J.; Maier-Wohlfart, S.; et al. Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update. Genes 2022, 13, 2327. https://doi.org/10.3390/genes13122327
Peschel N, Wright JT, Koster MI, Clarke AJ, Tadini G, Fete M, Hadj-Rabia S, Sybert VP, Norderyd J, Maier-Wohlfart S, et al. Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update. Genes. 2022; 13(12):2327. https://doi.org/10.3390/genes13122327
Chicago/Turabian StylePeschel, Nicolai, John T. Wright, Maranke I. Koster, Angus J. Clarke, Gianluca Tadini, Mary Fete, Smail Hadj-Rabia, Virginia P. Sybert, Johanna Norderyd, Sigrun Maier-Wohlfart, and et al. 2022. "Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update" Genes 13, no. 12: 2327. https://doi.org/10.3390/genes13122327
APA StylePeschel, N., Wright, J. T., Koster, M. I., Clarke, A. J., Tadini, G., Fete, M., Hadj-Rabia, S., Sybert, V. P., Norderyd, J., Maier-Wohlfart, S., Fete, T. J., Pagnan, N., Visinoni, A. F., & Schneider, H. (2022). Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update. Genes, 13(12), 2327. https://doi.org/10.3390/genes13122327