
Exploration of the Potential Transcriptional Regulatory Mechanisms of DNA Methyltransferases and MBD Genes in Petunia Anther Development and Multi-Stress Responses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of C5-MTase and MBD Genes
2.2. Phylogenetic and Gene Structure Analysis
2.3. Plant Materials and Stress Treatments
2.4. Isolation of RNA, Real-Time PCR, and Gene Expression Analyses
2.5. Subcellular Localization
3. Results
3.1. Identification of PhC5-MTase and PhMBD Genes
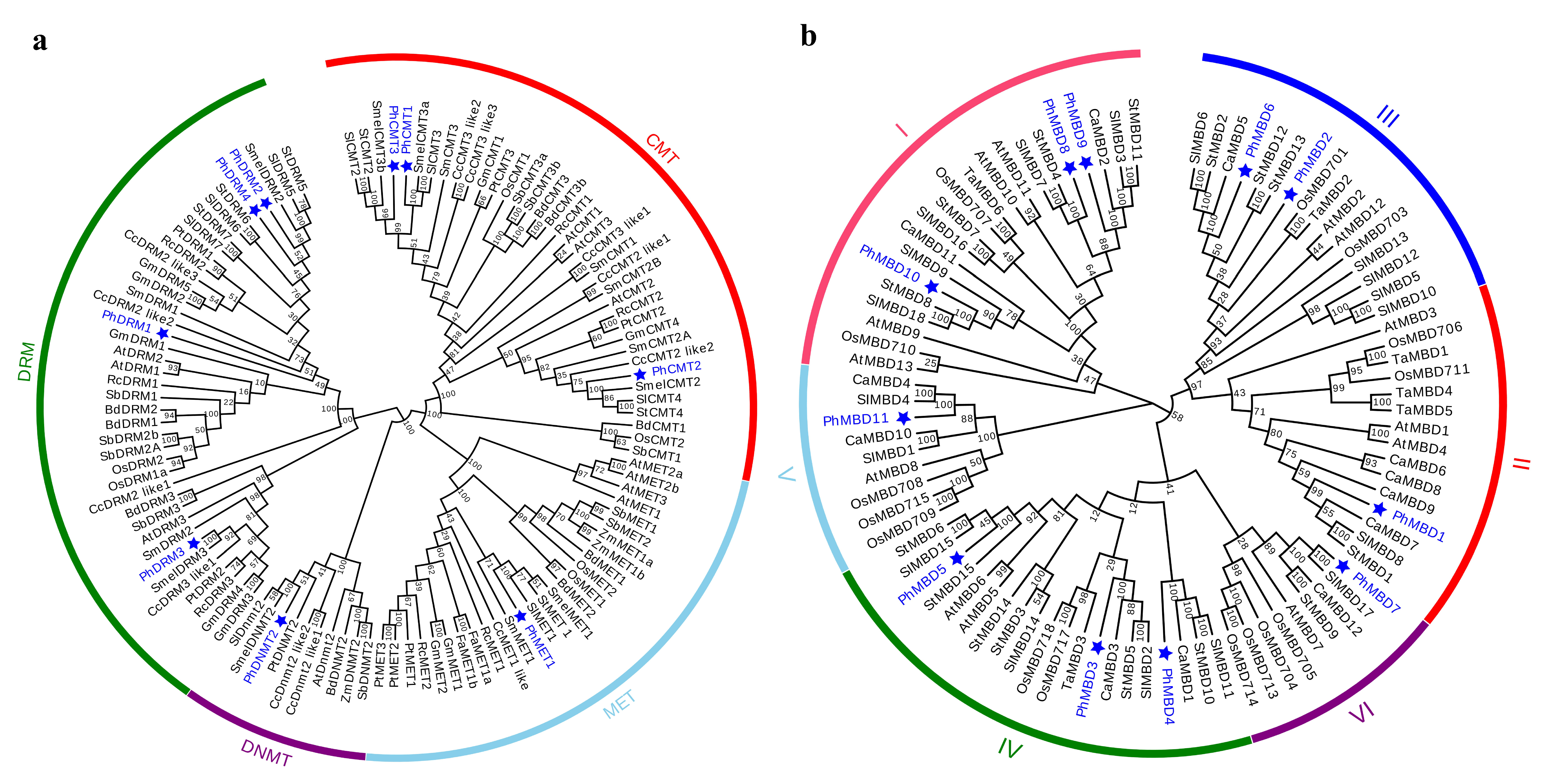
3.2. Phylogenetic Analysis and Classification
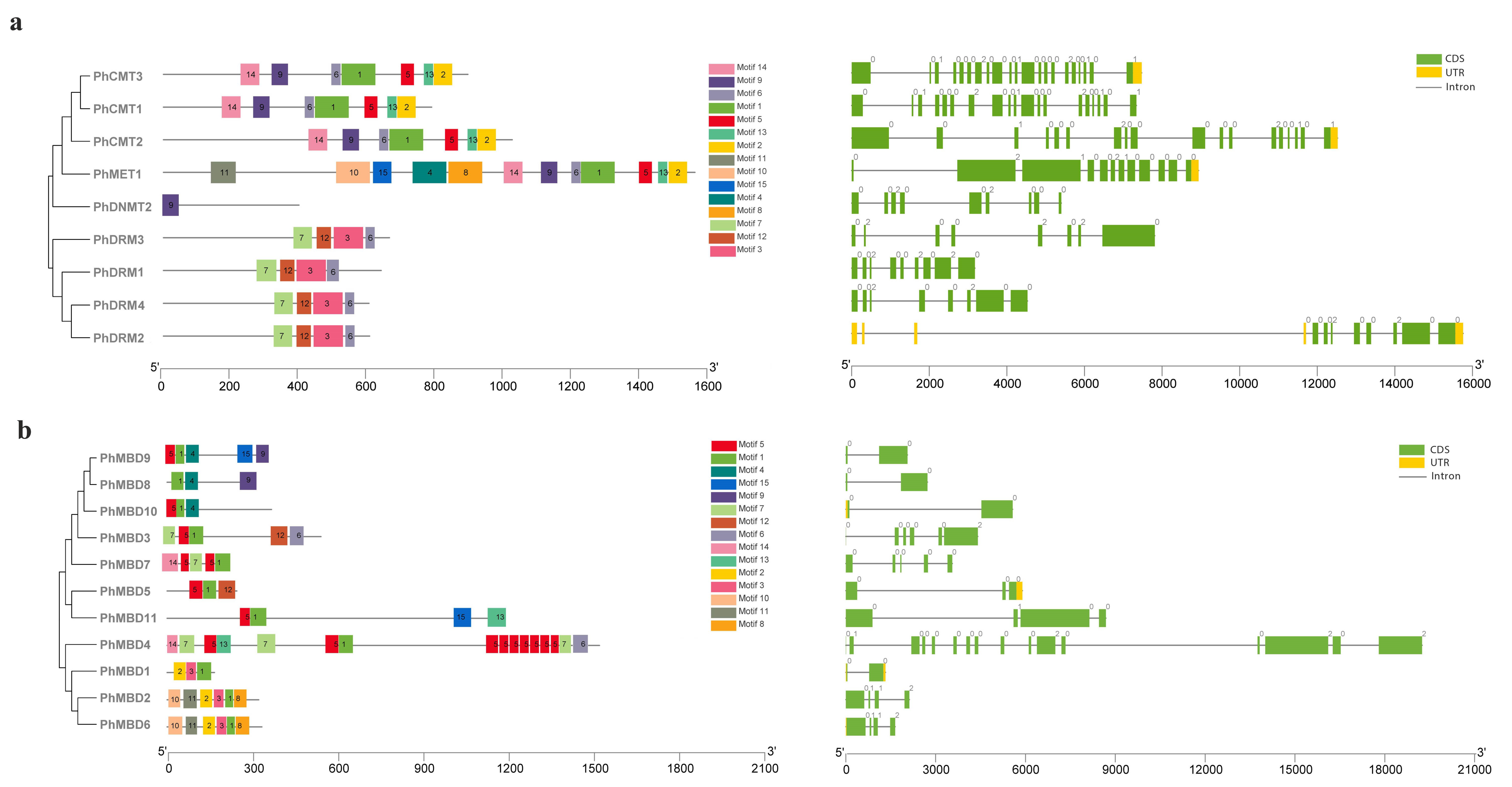
3.3. Multiple-Sequence Alignment and Motif Analysis
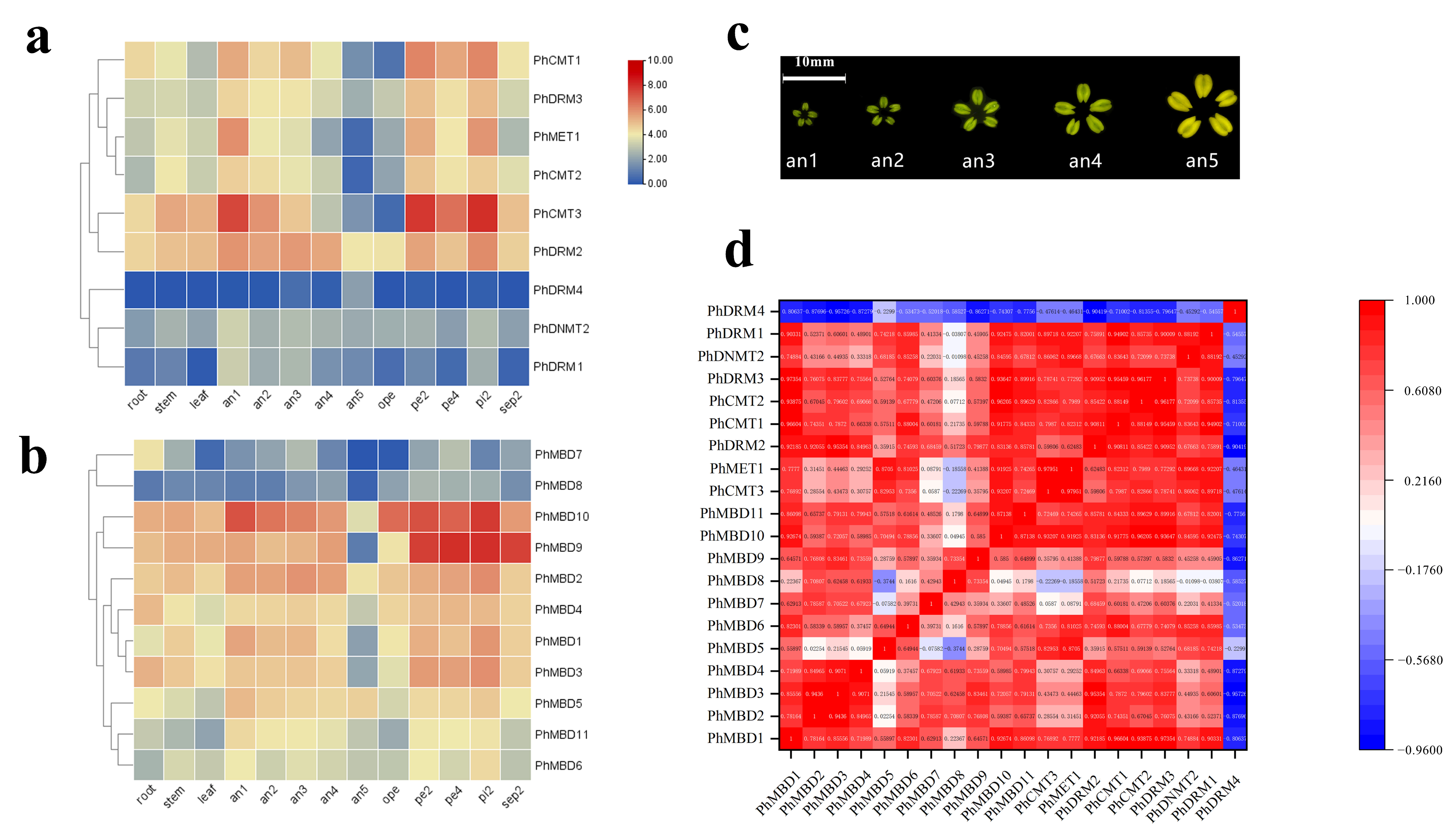
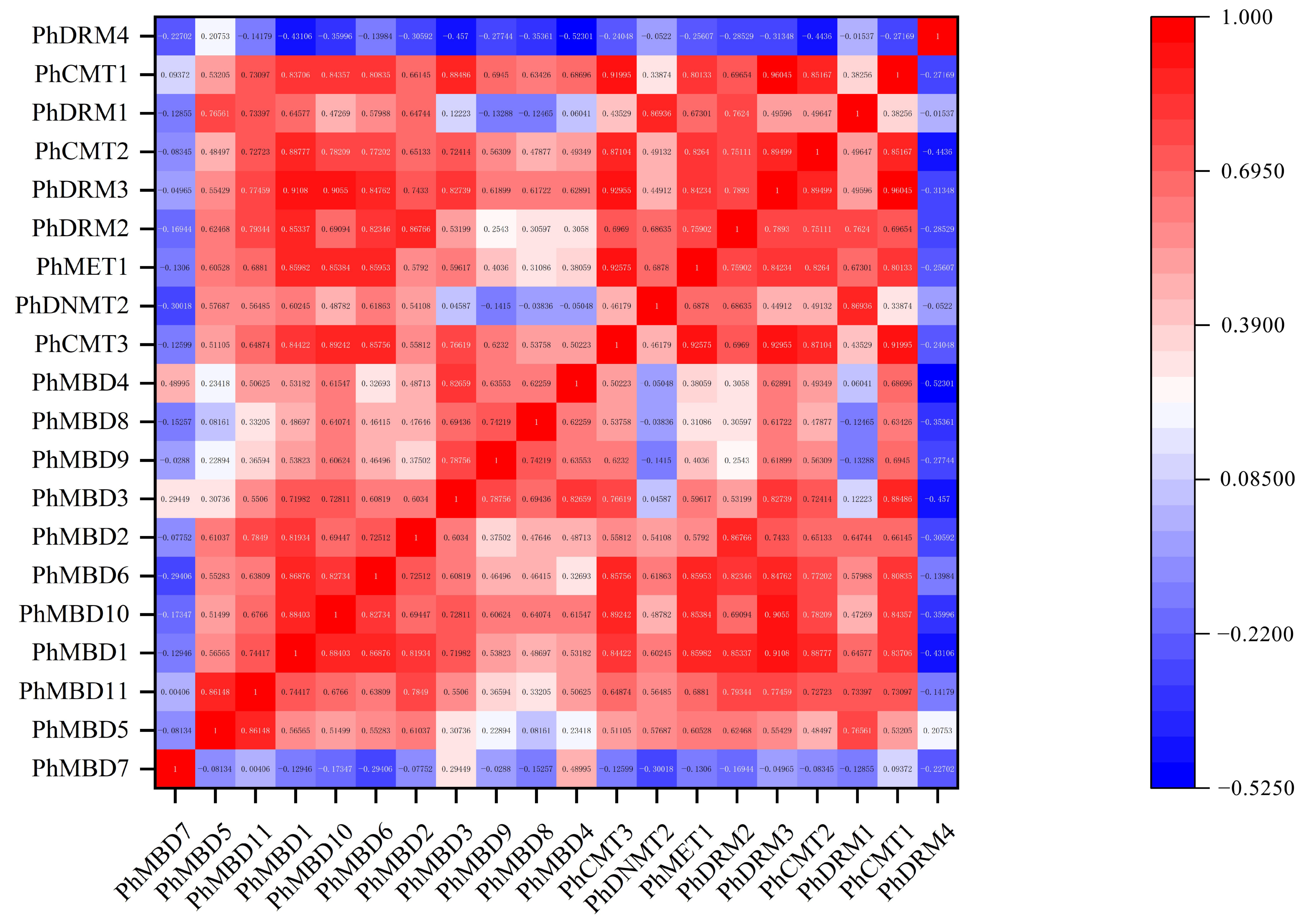
3.4. Expression Analysis of PhC5-MTases and PhMBDs
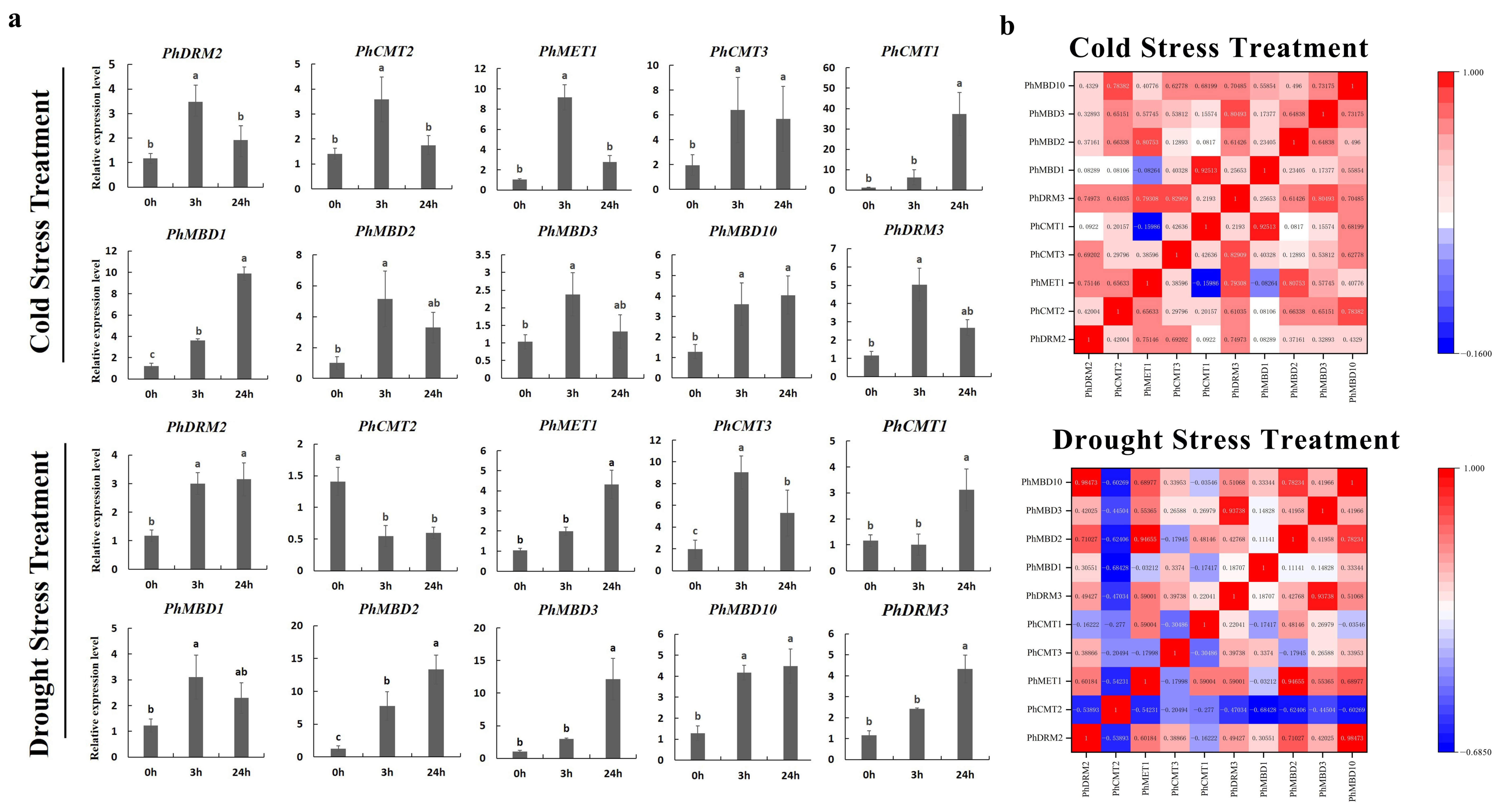
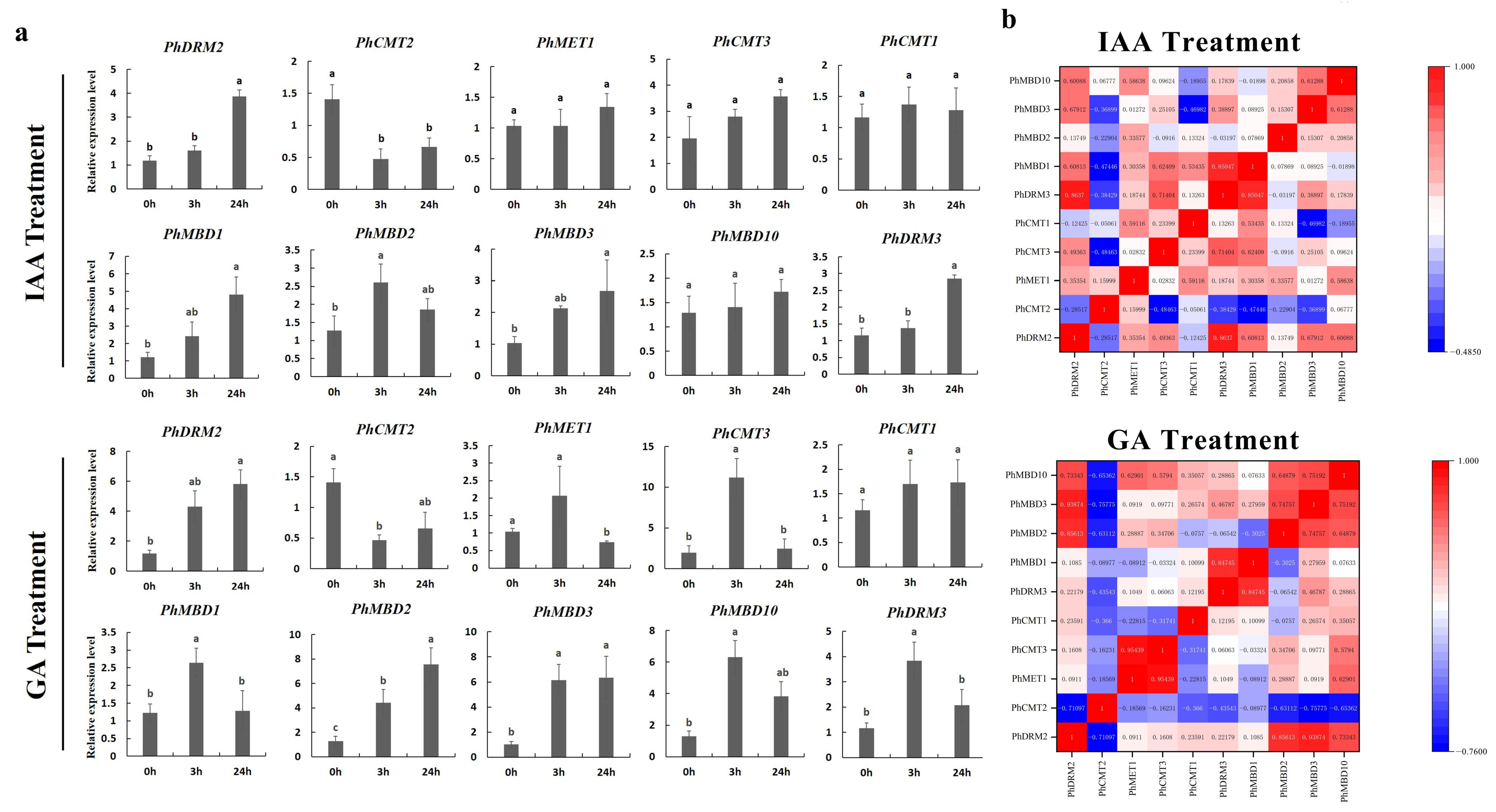
3.5. Expression Analysis of Phc5-Mtases and Phmbds in Response to Abiotic and Hormonal Treatments
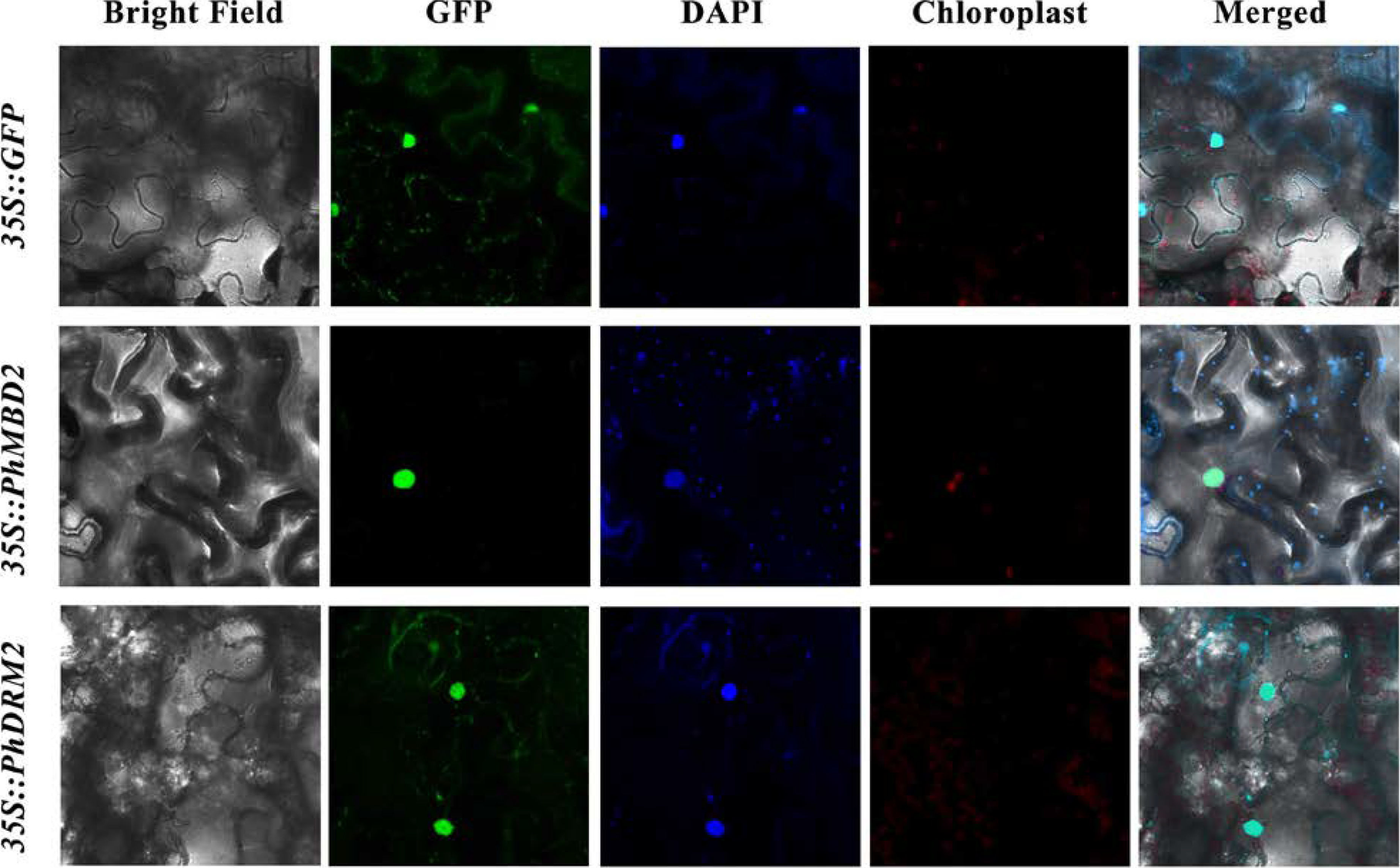
3.6. Subcellular Localization of PhDRM2 and PhMBD2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hirsch, S.; Baumberger, R.; Grossniklaus, U. Epigenetic variation, inheritance, and selection in plant populations. Cold Spring Harbor Symp. Quant. Biol. 2012, 77, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Luan, Y.; Chen, K.; Liu, Y.; Xiao, C.; Xie, Z. MethSMRT: An integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 2017, 45, D85–D89. [Google Scholar] [CrossRef] [Green Version]
- Bender, J. DNA methylation and epigenetics. Ann. Rev. Plant Biol. 2004, 55, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gao, C.; Bian, X.; Zhao, S.; Zhao, C.; Xia, H.; Song, H.; Hou, L.; Wan, S.; Wang, X. Genome-wide identification and comparative analysis of cytosine-5 DNA methyltransferase and demethylase families in wild and cultivated Peanut. Front. Plant Sci. 2016, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Grafi, G. Methyl-CpG-binding domain proteins in plants: Interpreters of DNA methylation. Trends Plant Sci. 2007, 12, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, Y.; Zhong, Q.; Zhan, K.; Bi, J.; Tang, J.; Xie, J.; Li, B. Identification and functional characterization of methyl-CpG binding domain protein from Tribolium Castaneum. Genomics 2020, 112, 2223–2232. [Google Scholar] [CrossRef]
- Grafi, G.; Zemach, A.; Pitto, L. Methyl-CpG-binding domain (MBD) proteins in plants. Biochim. Biophys. Acta Gene Struct. Expr. 2007, 1769, 287–294. [Google Scholar] [CrossRef]
- Springer, N.M.; Kaeppler, S.M. Evolutionary divergence of monocot and dicot Methyl-CpG-dinding domain proteins. Plant Physiol. 2005, 138, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Parida, A.P.; Raghuvanshi, U.; Pareek, A.; Singh, V.; Kumar, R.; Sharma, A.K. Genome-wide analysis of genes encoding MBD domain-containing proteins from tomato suggest their role in fruit development and abiotic stress responses. Mol. Biol. Rep. 2018, 45, 2653–2669. [Google Scholar] [CrossRef]
- Berg, A.; Meza, T.J.; Mahić, M.; Thorstensen, T.; Kristiansen, K.; Aalen, R.B. cgene family encoding methyl-CpG-binding domain proteins are transcriptionally active and at least one, AtMBD11, is crucial for normal development. Nucleic Acids Res. 2003, 31, 5291–5304. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Feng, Y.; Sun, J.; Zhang, W.; Han, Z.; Yu, S.; Gu, Y.; Cheng, X.; Lin, Z.; Na, M. Methyl-CpG-binding domain protein 3 promotes seizures by recruiting Methyltransferase DNMT1 to enhance TREM2 methylation. Neurochem. Res. 2021, 46, 2451–2462. [Google Scholar] [CrossRef]
- Li, J.; Deng, Q.; Fan, W.; Zeng, Q.; He, H.; Huang, F. Melatonin-induced suppression of DNA methylation promotes odontogenic differentiation in human dental pulp cells. Bioengineered 2020, 11, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Guan, X.; An, H.; Baihetiya, A.; Wang, W.; Shao, W.; Yang, H.; Wang, Y. Epigenetic silencing of miR-137 induces resistance to bicalutamide by targeting TRIM24 in prostate cancer cells. Am. J. Transl. Res. 2019, 11, 3226–3237. [Google Scholar] [PubMed]
- Chatraryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2016, 45, D369–D379. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.T.S.; Inzé, D.; Nelissen, H. Differential methylation during maize leaf growth targets developmentally regulated genes. Plant Physiol. 2014, 164, 1350–1364. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Ju, Z.; Gao, C.; Mei, X.; Fu, D.; Zhu, H.; Luo, Y.; Zhu, B. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum Lycopersicum. Gene 2014, 550, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Zhang, Y.; Zhou, S.; Hu, W.; Wu, X.; Ye, Y.; Wu, X.; Xiao, Y.; Li, X.; Xue, H. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnegan, E.J.; Kovac, K.A.; Jaligot, E.; Sheldon, C.C.; James Peacock, W.; Dennis, E.S. The downregulation of FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA methylation and by vernalization occurs by distinct mechanisms. Plant J. Cell Mol. Biol. 2005, 44, 420–432. [Google Scholar] [CrossRef]
- Goto, N.; Pharis, R.P. Role of gibberellins in the development of floral organs of the gibberellin-deficient mutant, ga1-1, of Arabidopsis Thaliana. Can. J. Bot. 1999, 77, 944–954. [Google Scholar]
- Hirano, K.; Aya, K.; Hobo, T.; Sakakibara, H.; Kojima, M.; Shim, R.A.; Hasegawa, Y.; Ueguchi-Tanaka, M.; Matsuoka, M. Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in microspore/pollen and tapetum of rice. Plant Cell Physiol. 2008, 49, 1429–1450. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B. Enzymatic DNA methylation is an epigenetic control for genetic functions of the cell. Biochemistry 2005, 70, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Solís, M.-T.; Chakrabarti, N.; Corredor, E.; Cortés-Eslava, J.; Rodríguez-Serrano, M.; Biggiogera, M.; Risueño, M.C.; Testillano, P.S. Epigenetic changes accompany developmental programmed cell death in tapetum cells. Plant Cell Physiol. 2014, 55, 16–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forino, L.M.C.; Andreucci, A.C.; Tredici, I.D.; Felici, C.; Giraldi, E.; Tagliasacchi, A.M. DNA methylation of tapetum cells during microsporogenesis in Malus domestica Borkh. Isr. J. Plant Sci. 2003, 51, 91–100. [Google Scholar] [CrossRef]
- Qian, Y.; Ren, Q.; Jiang, L.; Zhang, J.; Chen, C.; Chen, L. Genome-wide analysis of maize MBD gene family and expression profiling under abiotic stress treatment at the seedling stage. Plant Biotechnol. Rep. 2020, 14, 323–338. [Google Scholar] [CrossRef]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Ogneva, Z.V.; Dubrovina, A.S.; Kiselev, K.V. Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis Thaliana. Biol. Plant 2016, 60, 628–634. [Google Scholar] [CrossRef]
- Qian, Y.; Xi, Y.; Cheng, B.; Zhu, S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014, 33, 1661–1672. [Google Scholar] [CrossRef]
- Gahlaut, V.; Samtani, H.; Khurana, P. Genome-wide identification and expression profiling of cytosine-5 DNA methyltransferases during drought and heat stress in wheat (Triticum aestivum). Genomics 2020, 112, 4796–4807. [Google Scholar] [CrossRef]
- Li, Y.; Meng, F.; Yin, J.; Liu, H.; Si, Z.; Ni, Z.; Sun, Q.; Ren, J.; Niu, H. Isolation and comparative expression analysis of six MBD genes in wheat. Biochim. Biophys. Acta Gene Regul. Mech 2008, 1779, 90–98. [Google Scholar] [CrossRef]
- Bombarely, A.; Moser, M.; Amrad, A.; Bao, M.; Bapaume, L.; Barry, C.S.; Bliek, M.; Boersma, M.R.; Borghi, L.; Bruggmann, R.; et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia Hybrida. Nat. Plants 2016, 2, 16074. [Google Scholar] [CrossRef] [Green Version]
- Vandenbussche, M.; Chambrier, P.; Rodrigues Bento, S.; Morel, P. Petunia, your next supermodel? Front. Plant Sci. 2016, 7, 72. [Google Scholar] [CrossRef]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden markov model speed heuristic and iterative HMM search procedure. BMC Bioinf. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2014, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- Ali, F.; Qanmber, G.; Wei, Z.; Yu, D.; Li, Y.h.; Gan, L.; Li, F.; Wang, Z. Genome-wide characterization and expression analysis of geranylgeranyl diphosphate synthase genes in cotton (Gossypium spp.) in plant development and abiotic stresses. BMC Genom. 2020, 21, 561. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Jing, X.; Chen, Y.; Hu, F. Functional analysis of CgWRKY57 from Cymbidium goeringii in ABA response. PeerJ 2021, 9, e10982. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Yin, C.; Guo, R.; Peng, H.; Yang, Z.; Liu, G.; Bao, M.; Hu, H. An anther-specific gene PhGRP is regulated by PhMYC2 and causes male sterility when overexpressed in petunia anthers. Plant Cell Rep. 2017, 36, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, T.; Wang, M.; Li, T.; Wang, G.; Fu, F.-F.; Cao, F. Systematic investigation and expression profiles of the GbR2R3-MYB transcription factor family in ginkgo (Ginkgo biloba L.). Int. J. Biol. Macromol. 2021, 172, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Du, J.; Li, Y.; Thomas, H.R.; Frank, M.H.; Wang, L.; Hu, H. Insight into the petunia Dof transcription factor family reveals a new regulator of male-sterility. Ind. Crops Prod. 2021, 161, 113196. [Google Scholar] [CrossRef]
- Lalitha, S. Primer premier 5. Biotech. Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Jeltsch, A. Molecular enzymology of mammalian DNA methyltransferases. In DNA Methylation: Basic Mechanisms; Doerfler, W., Böhm, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 203–225. [Google Scholar]
- Pavlopoulou, A.; Kossida, S. Plant cytosine-5 DNA methyltransferases: Structure, function, and molecular evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Callebaut, I.; Courvalin, J.-C.; Mornon, J.-P. The BAH (bromo-adjacent homology) domain: A link between DNA methylation, replication and transcriptional regulation. FEBS Lett. 1999, 446, 189–193. [Google Scholar] [CrossRef]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Liu, G.; Xia, Y.; Liu, T.; Dai, S.; Hou, X. The DNA methylome and association of differentially methylated regions with differential gene expression during heat stress in Brassica rapa. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; Zhang, L.; Li, J.; Shen, C.; Qiu, P.; Tu, L.; Zhang, X.; Wang, M. Tracing the origin and evolution history of methylation-related genes in plants. BMC Plant Biol. 2019, 19, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moglia, A.; Gianoglio, S.; Acquadro, A.; Valentino, D.; Milani, A.M.; Lanteri, S.; Comino, C. Identification of DNA methyltransferases and demethylases in Solanum melongena L., and their transcription dynamics during fruit development and after salt and drought stresses. PLoS ONE 2019, 14, e0223581. [Google Scholar] [CrossRef] [Green Version]
- Gu, T.; Ren, S.; Wang, Y.; Han, Y.; Li, Y. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca. Mol. Genet. Genom. 2016, 291, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steward, N.; Ito, M.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Periodic DNA methylation in Maize nucleosomes and demethylation by environmental stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Zhang, S.; Zhou, C.; Chen, L.; Fu, H.; Li, X.; Lin, Y.; Lai, Z.; Guo, Y. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PeerJ 2020, 8, e8432. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Chauhan, P.K.; Khurana, A. Identification and expression profiling of DNA methyltransferases during development and stress conditions in Solanaceae. Funct. Integr. Genom. 2016, 16, 513–528. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Shen, H.; Liu, J.; Hu, H.; Tan, H.; Yang, X.; Wang, L.; Yue, Y. Exploration of the Potential Transcriptional Regulatory Mechanisms of DNA Methyltransferases and MBD Genes in Petunia Anther Development and Multi-Stress Responses. Genes 2022, 13, 314. https://doi.org/10.3390/genes13020314
Shi L, Shen H, Liu J, Hu H, Tan H, Yang X, Wang L, Yue Y. Exploration of the Potential Transcriptional Regulatory Mechanisms of DNA Methyltransferases and MBD Genes in Petunia Anther Development and Multi-Stress Responses. Genes. 2022; 13(2):314. https://doi.org/10.3390/genes13020314
Chicago/Turabian StyleShi, Lisha, Huimin Shen, Jiawei Liu, Hongmin Hu, Hongyan Tan, Xiulian Yang, Lianggui Wang, and Yuanzheng Yue. 2022. "Exploration of the Potential Transcriptional Regulatory Mechanisms of DNA Methyltransferases and MBD Genes in Petunia Anther Development and Multi-Stress Responses" Genes 13, no. 2: 314. https://doi.org/10.3390/genes13020314
APA StyleShi, L., Shen, H., Liu, J., Hu, H., Tan, H., Yang, X., Wang, L., & Yue, Y. (2022). Exploration of the Potential Transcriptional Regulatory Mechanisms of DNA Methyltransferases and MBD Genes in Petunia Anther Development and Multi-Stress Responses. Genes, 13(2), 314. https://doi.org/10.3390/genes13020314