
Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child

 , , , , and
, , , , and 
Abstract
:1. Introduction
1.1. Background
1.2. MPS-PS
1.3. Pathogenesis and Genetic Analysis
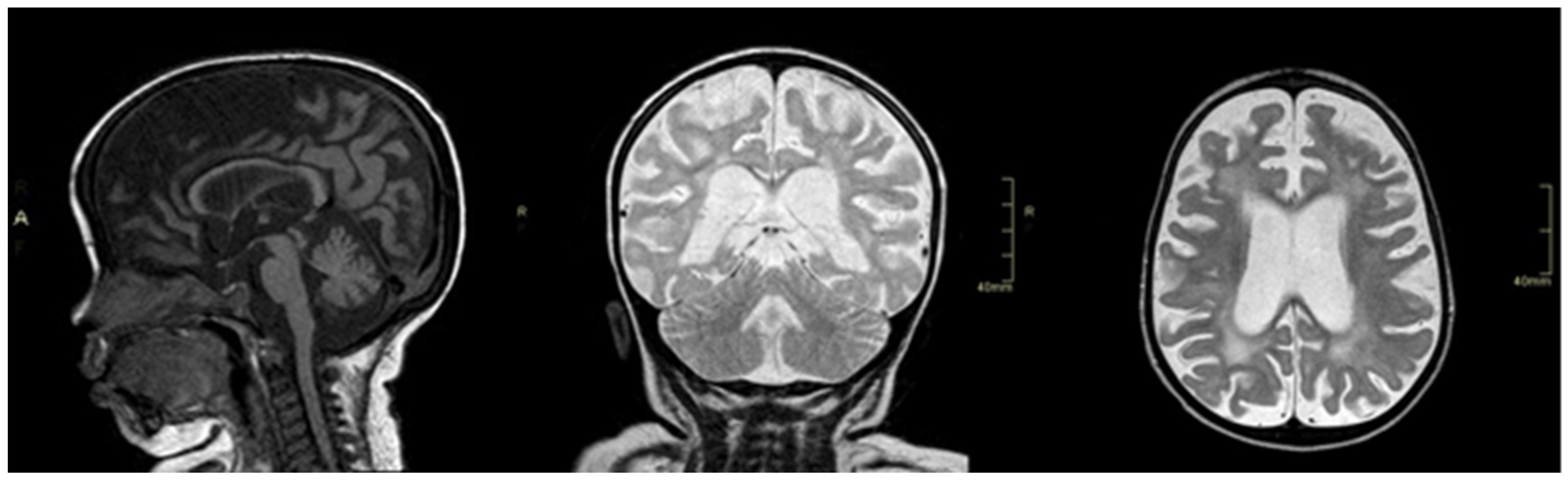
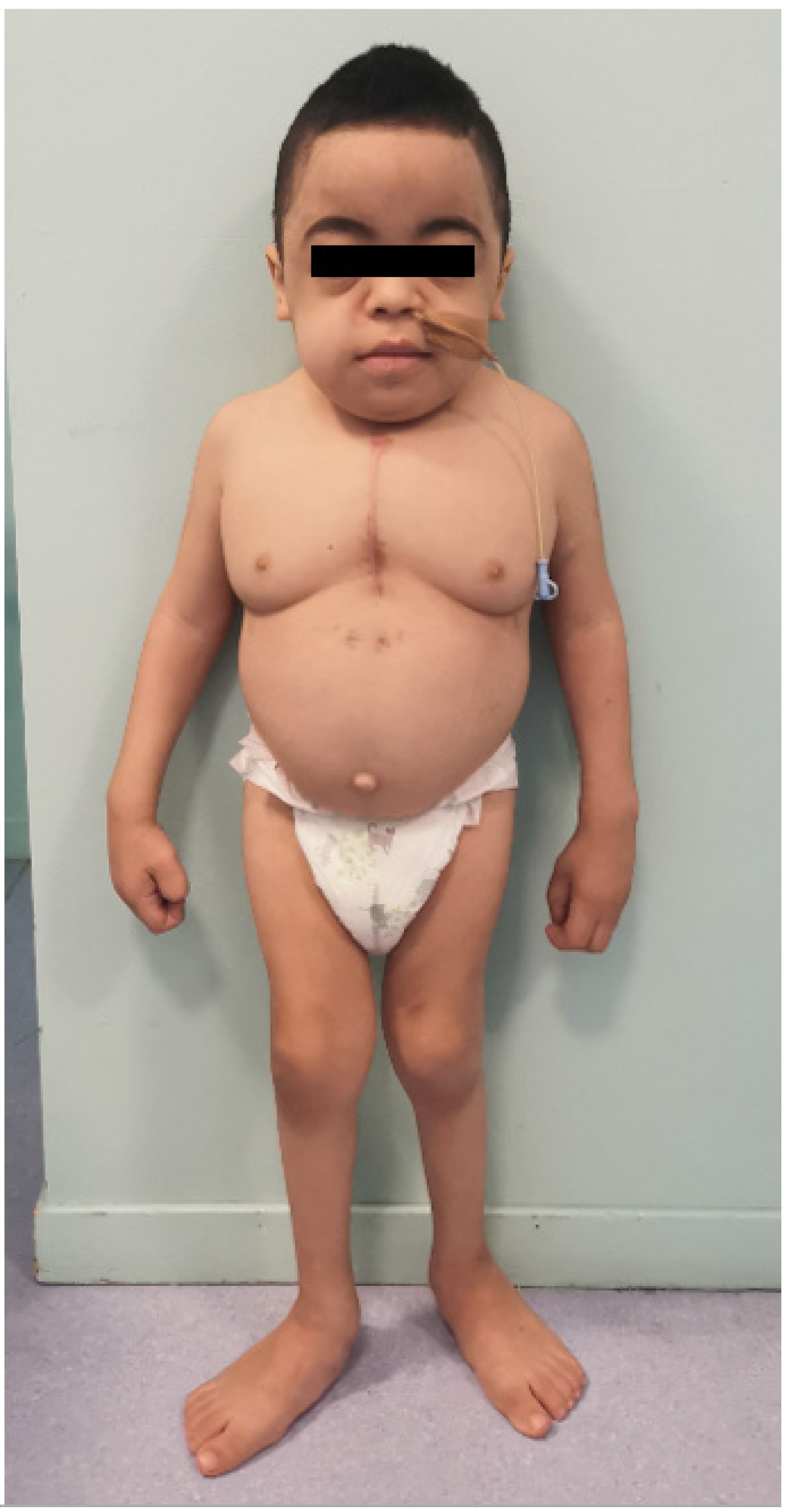
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Guerrero, J.L.; Higuera, P.J.I.G.; Flórez, J.S.A.; Contreras-García, G.A. Mucopolisacaridosis: Características clínicas, diagnóstico y de manejo. Rev. Chil. Pediatr. 2016, 87, 295–304. [Google Scholar] [CrossRef]
- Sawamoto, K.; Chen, H.-H.; Alméciga-Díaz, C.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 123, 59–68. [Google Scholar] [CrossRef]
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; De Pellegrin, M.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation inVPS33Aaffects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 2016, 26, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, E.V.; Shatunov, A.; Wartosch, L.; I Moskvina, A.; E Nikolaeva, L.; A Bright, N.; Tylee, K.L.; Church, H.J.; Ballabio, A.; Luzio, J.P.; et al. The lysosomal disease caused by mutant VPS33A. Hum. Mol. Genet. 2019, 28, 2514–2530. [Google Scholar] [CrossRef]
- Dursun, A.; Yalnizoglu, D.; Gerdan, O.F.; Yucel-Yilmaz, D.; Sagiroglu, M.S.; Yuksel, B.; Gucer, S.; Sivri, S.; Ozgul, R.K. A probable new syndrome with the storage disease phenotype caused by the VPS33A gene mutation. Clin. Dysmorphol. 2017, 26, 1–12. [Google Scholar] [CrossRef]
- Tarskaia, L.A.; Zinchenko, R.A.; Elchinova, G.I.; Egorova, A.G.; Korotov, M.N.; Basova, E.V.; Prokopeva, A.M.; Sivtseva, E.N.; Nikolaeva, E.E.; Banshikova, E.S.; et al. The Structure and Diversity of Hereditary Pathology in Sakha Republic (Yakutia). Russ. J. Genet. 2004, 40, 1264–1272. [Google Scholar] [CrossRef]
- Maksimova, N.; Hara, K.; Miyashia, A.; Nikolaeva, I.; Shiga, A.; Nogovicina, A.; Sukhomyasova, A.; Argunov, V.; Shvedova, A.; Ikeuchi, T.; et al. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: New population isolate in Asia. J. Med. Genet. 2007, 44, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Pakendorf, B.; Novgorodov, I.N.; Osakovskij, V.L.; Danilova, A.P.; Protod’Jakonov, A.P.; Stoneking, M. Investigating the effects of prehistoric migrations in Siberia: Genetic variation and the origins of Yakuts. Qual. Life Res. 2006, 120, 334–353. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, S.; Khusnutdinova, E. Gene pools of peoples from the Republic of Sakha (Yakutiia): Structure, origin, genetic relationships. Genetika 2010, 46, 1244–1246. [Google Scholar]
- Solinger, J.A.; Spang, A. Tethering complexes in the endocytic pathway: CORVET and HOPS. FEBS J. 2013, 280, 2743–2757. [Google Scholar] [CrossRef]
- Raymond, C.K.; Howald-Stevenson, I.; A Vater, C.; Stevens, T.H. Morphological classification of the yeast vacuolar protein sorting mutants: Evidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell 1992, 3, 1389–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rappard, D.F.; Boelens, J.J.; Wolf, N.I. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Adang, L.A.; Radhakrishnan, K.; Ahrens-Nicklas, R.C. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int. J. Mol. Sci. 2020, 21, 3448. [Google Scholar] [CrossRef]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and Uncommon Pathogenic Cascades in Lysosomal Storage Diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fésüs, L.; Demény, M.A.; Petrovski, G. Autophagy Shapes Inflammation. Antioxid. Redox Signal. 2011, 14, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; Van Der Spoel, A.C.; D’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Elder, M.E.; Nayak, S.; Collins, S.W.; Lawson, L.A.; Kelley, J.S.; Herzog, R.W.; Modica, R.F.; Lew, J.; Lawrence, R.M.; Byrne, B.J. B-Cell Depletion and Immunomodulation before Initiation of Enzyme Replacement Therapy Blocks the Immune Response to Acid Alpha-Glucosidase in Infantile-Onset Pompe Disease. J. Pediatr. 2013, 163, 847–854.e1. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Month | Steroid | Dosage | Dosage/Square Metre Per Day |
|---|---|---|---|
| Month 1 | DXM | 1.4 + 1.4 mg | 6 mg/sqm |
| Month 3 | DXM | 1.3 + 1.3 mg | 5 mg/sqm |
| Month 5 | DXM | 1.2 + 1.2 mg | 4.7 mg/sqm |
| Month 6 | DXM | 1.1 + 1.1 mg | 4.1 mg/sqm |
| Month 6 | DXM | 1 + 1 mg | 3.7 mg/sqm |
| Month 8 | DXM | 0.9 + 0.9 mg | 3.2 mg/sqm |
| Month 9 | DXM | 0.8 + 0.8 mg | 2.8 mg/sqm |
| Month 10 | DXM | 0.5 + 0.5 mg | 1.8 mg/sqm |
| Month 11 | HC | 10 + 10 + 10 mg | 53.6 mg/sqm |
| Month 12 | HC | 10 + 10 + 5 mg | 44 mg/sqm |
| Month 13 | HC | 10 + 10 mg | 35 mg/sqm |
| Month 20 | HC | 7.5 + 7.5 mg | 26.8 mg/sqm |
| Month 23 | HC | 5 + 5 mg | 19 mg/sqm |
| Month 30 Ongoing | HC | 5 + 2.5 mg | 12.5 mg/sqm |
| Month 42 | Synachten test | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faraguna, M.C.; Musto, F.; Crescitelli, V.; Iascone, M.; Spaccini, L.; Tonduti, D.; Fedeli, T.; Kullmann, G.; Canonico, F.; Cattoni, A.; et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes 2022, 13, 442. https://doi.org/10.3390/genes13030442
Faraguna MC, Musto F, Crescitelli V, Iascone M, Spaccini L, Tonduti D, Fedeli T, Kullmann G, Canonico F, Cattoni A, et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes. 2022; 13(3):442. https://doi.org/10.3390/genes13030442
Chicago/Turabian StyleFaraguna, Martha Caterina, Francesca Musto, Viola Crescitelli, Maria Iascone, Luigina Spaccini, Davide Tonduti, Tiziana Fedeli, Gaia Kullmann, Francesco Canonico, Alessandro Cattoni, and et al. 2022. "Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child" Genes 13, no. 3: 442. https://doi.org/10.3390/genes13030442
APA StyleFaraguna, M. C., Musto, F., Crescitelli, V., Iascone, M., Spaccini, L., Tonduti, D., Fedeli, T., Kullmann, G., Canonico, F., Cattoni, A., Dell’Acqua, F., Rizzari, C., & Gasperini, S. (2022). Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes, 13(3), 442. https://doi.org/10.3390/genes13030442