
The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism



Abstract
:1. The Ehlers–Danlos Syndromes, a General Introduction
2. The Clinical Presentation of EDS, an Overview
2.1. Prenatal Findings
2.2. Integumentary System
2.3. Skeletal System
2.4. Neuromuscular System
2.5. Cardiovascular System
2.6. Ocular System
2.7. Orocraniofacial
2.8. Other
3. The Genetic and Pathophysiological Basis of the EDS
3.1. Defects in Fibrillar Collagen Structure and Processing
3.2. Defects in Collagen Crosslinking and Folding
3.3. Defects in ECM Bridging Molecules
3.4. Glycosaminoglycan Biosynthesis
3.5. Defects in Intracellular Processes
3.6. Defects in the Complement Pathway
4. Diagnostic Approach
4.1. Biochemical Analyses
4.2. Ultrastructural Collagen Fibril Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers–Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L. The structure of the connective tissue, an explanation of the symptoms of the Ehlers-Danlos syndrome. Dermatology 1955, 110, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; de Paepe, A.; Danks, D.; Finidori, G.; Gedde-Dahl, T.; Goodman, R.; Hall, J.G.; Hollister, D.W.; Horton, W.; McKusick, V.A.; et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am. J. Med. Genet. 1988, 29, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, P.; Regier, D. Inborn Errors of Metabolism: From Preconception to Adulthood. Am. Fam. Physician 2019, 99, 25–32. [Google Scholar]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, T.; Colige, A.; Syx, D.; Giunta, C.; Lindert, U.; Rohrbach, M.; Aryani, O.; Alanay, Y.; Simsek-Kiper, P.O.; Kroes, H.Y.; et al. Expanding the clinical and mutational spectrum of the Ehlers-Danlos syndrome, dermatosparaxis type. Genet. Med. 2016, 18, 882–891. [Google Scholar] [CrossRef] [Green Version]
- D’Hondt, S.; Van Damme, T.; Malfait, F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: A systematic review. Genet. Med. 2018, 20, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rymen, D.; Jaeken, J. Skin manifestations in CDG. J. Inherit. Metab. Dis. 2014, 37, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Remvig, L.; Jensen, D.V.; Ward, R.C. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: Review of the literature. J. Rheumatol. 2007, 34, 804–809. [Google Scholar] [PubMed]
- Castiglioni, C.; Feillet, F.; Barnerias, C.; Wiedemann, A.; Muchart, J.; Cortes, F.; Hernando-Davalillo, C.; Montero, R.; Dupré, T.; Bruneel, A.; et al. Expanding the phenotype of X-linked SSR4-CDG: Connective tissue implications. Hum. Mutat. 2021, 42, 142–149. [Google Scholar] [CrossRef]
- Malfait, F.; Colman, M.; Vroman, R.; De Wandele, I.; Rombaut, L.; Miller, R.E.; Malfait, A.M.; Syx, D. Pain in the Ehlers-Danlos syndromes: Mechanisms, models, and challenges. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 429–445. [Google Scholar] [CrossRef]
- Ayoub, S.; Ghali, N.; Angwin, C.; Baker, D.; Baffini, S.; Brady, A.F.; Giovannucci Uzielli, M.L.; Giunta, C.; Johnson, D.S.; Kosho, T.; et al. Clinical features, molecular results, and management of 12 individuals with the rare arthrochalasia Ehlers-Danlos syndrome. Am. J. Med. Genet. A 2020, 182, 994–1007. [Google Scholar] [CrossRef]
- Van Damme, T.; Pang, X.; Guillemyn, B.; Gulberti, S.; Syx, D.; De Rycke, R.; Kaye, O.; de Die-Smulders, C.E.M.; Pfundt, R.; Kariminejad, A.; et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers-Danlos syndrome. Hum. Mol. Genet. 2018, 27, 3475–3487. [Google Scholar] [CrossRef] [Green Version]
- Hicks, D.; Farsani, G.T.; Laval, S.; Collins, J.; Sarkozy, A.; Martoni, E.; Shah, A.; Zou, Y.; Koch, M.; Bönnemann, C.G.; et al. Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy. Hum. Mol. Genet. 2014, 23, 2353–2363. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zwolanek, D.; Izu, Y.; Gandhy, S.; Schreiber, G.; Brockmann, K.; Devoto, M.; Tian, Z.; Hu, Y.; Veit, G.; et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum. Mol. Genet. 2014, 23, 2339–2352. [Google Scholar] [CrossRef] [Green Version]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voermans, N.C.; Kempers, M.; Lammens, M.; van Alfen, N.; Janssen, M.C.; Bonnemann, C.; van Engelen, B.G.; Hamel, B.C. Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. A 2012, 158, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. EJHG 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, M.R.; Lee, C.L.; Lin, S.M.; Hung, C.L.; Niu, D.M.; Chang, T.M.; Chuang, C.K.; Lin, S.P. Aortic Root Dilatation in Taiwanese Patients with Mucopolysaccharidoses and the Long-Term Effects of Enzyme Replacement Therapy. Diagnostics 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Symoens, S.; Coucke, P.; Nunes, L.; De Almeida, S.; De Paepe, A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J. Med. Genet. 2006, 43, e36. [Google Scholar] [CrossRef] [PubMed]
- Dhooge, T.; Van Damme, T.; Syx, D.; Mosquera, L.M.; Nampoothiri, S.; Radhakrishnan, A.; Simsek-Kiper, P.O.; Utine, G.E.; Bonduelle, M.; Migeotte, I.; et al. More than meets the eye: Expanding and reviewing the clinical and mutational spectrum of brittle cornea syndrome. Hum. Mutat. 2021, 42, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Kapferer-Seebacher, I.; Pepin, M.; Werner, R.; Aitman, T.J.; Nordgren, A.; Stoiber, H.; Thielens, N.; Gaboriaud, C.; Amberger, A.; Schossig, A.; et al. Periodontal Ehlers-Danlos Syndrome Is Caused by Mutations in C1R and C1S, which Encode Subcomponents C1r and C1s of Complement. Am. J. Hum. Genet. 2016, 99, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Birk, D.E.; Brückner, P. Collagens, Suprastructures, and Collagen Fibril Assembly. In The Extracellular Matrix: An Overview; Springer: Berlin/Heidelberg, Germany, 2011; pp. 77–115. [Google Scholar]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Birk, D.E. Type V collagen: Heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 2001, 32, 223–237. [Google Scholar] [CrossRef]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenspan, D.S. Biosynthetic processing of collagen molecules. Collagen 2005, 247, 149–183. [Google Scholar]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef] [Green Version]
- Zoppi, N.; Gardella, R.; De Paepe, A.; Barlati, S.; Colombi, M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate alpha2beta1 integrin, and recruit alphavbeta3 Instead of alpha5beta1 integrin. J. Biol. Chem. 2004, 279, 18157–18168. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS ONE 2019, 14, e0211647. [Google Scholar] [CrossRef] [Green Version]
- Keene, D.R.; Sakai, L.Y.; Bächinger, H.P.; Burgeson, R.E. Type III collagen can be present on banded collagen fibrils regardless of fibril diameter. J. Cell Biol. 1987, 105, 2393–2402. [Google Scholar] [CrossRef]
- Romanic, A.M.; Adachi, E.; Kadler, K.E.; Hojima, Y.; Prockop, D.J. Copolymerization of pNcollagen III and collagen I. pNcollagen III decreases the rate of incorporation of collagen I into fibrils, the amount of collagen I incorporated, and the diameter of the fibrils formed. J. Biol. Chem. 1991, 266, 12703–12709. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS ONE 2018, 13, e0191220. [Google Scholar] [CrossRef] [Green Version]
- Crowther, M.A.; Lach, B.; Dunmore, P.J.; Roach, M.R. Vascular collagen fibril morphology in type IV Ehlers-Danlos syndrome. Connect Tissue Res. 1991, 25, 209–217. [Google Scholar] [CrossRef]
- Byers, P.H.; Duvic, M.; Atkinson, M.; Robinow, M.; Smith, L.T.; Krane, S.M.; Greally, M.T.; Ludman, M.; Matalon, R.; Pauker, S.; et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 1997, 72, 94–105. [Google Scholar] [CrossRef]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Leduc, C.; Dupont, L.; Janssen, L.; Delolme, F.; Vadon-Le Goff, S.; Smargiasso, N.; Baiwir, D.; Mazzucchelli, G.; Zanella-Cleon, I.; et al. Determination of the substrate repertoire of ADAMTS2, 3, and 14 significantly broadens their functions and identifies extracellular matrix organization and TGF-β signaling as primary targets. FASEB J. 2016, 30, 1741–1756. [Google Scholar] [CrossRef] [Green Version]
- Giunta, C.; Chambaz, C.; Pedemonte, M.; Scapolan, S.; Steinmann, B. The arthrochalasia type of Ehlers-Danlos syndrome (EDS VIIA and VIIB): The diagnostic value of collagen fibril ultrastructure. Am. J. Med. Genet. A 2008, 146, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Symoens, S.; De Backer, J.; Hermanns-Le, T.; Sakalihasan, N.; Lapiere, C.M.; Coucke, P.; De Paepe, A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum. Mutat. 2007, 28, 387–395. [Google Scholar] [CrossRef]
- Nuytinck, L.; Freund, M.; Lagae, L.; Pierard, G.E.; Hermanns-Le, T.; De Paepe, A. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am. J. Hum. Genet. 2000, 66, 1398–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krane, S.M.; Pinnell, S.R.; Erbe, R.W. Lysyl-protocollagen hydroxylase deficiency in fibroblasts from siblings with hydroxylysine-deficient collagen. Proc. Natl. Acad. Sci. USA 1972, 69, 2899–2903. [Google Scholar] [CrossRef] [Green Version]
- Pinnell, S.R.; Krane, S.M.; Kenzora, J.E.; Glimcher, M.J. A heritable disorder of connective tissue: Hydroxylysine-deficient collagen disease. N. Engl. J. Med. 1972, 286, 1013–1020. [Google Scholar] [CrossRef]
- Hyland, J.; Ala-Kokko, L.; Royce, P.; Steinmann, B.; Kivirikko, K.I.; Myllylä, R. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers–Danlos syndrome type VI. Nat. Genet. 1992, 2, 228–231. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Bachinger, H.P. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J. Biol. Chem. 2014, 289, 18189–18201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Mizuno, K.; Bachinger, H.P. Ziploc-ing the structure 2.0: Endoplasmic reticulum-resident peptidyl prolyl isomerases show different activities toward hydroxyproline. J. Biol. Chem. 2017, 292, 9273–9282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Mizuno, N.; Holden, P.; Lim, P.J.; Gould, D.B.; Rohrbach, M.; Giunta, C.; Bächinger, H.P. The novel missense mutation Met48Lys in FKBP22 changes its structure and functions. Sci. Rep. 2020, 10, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.; Giunta, C.; Krabichler, B.; Ruschendorf, F.; Zoppi, N.; Colombi, M.; Bittner, R.E.; Quijano-Roy, S.; Muntoni, F.; Cirak, S.; et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 2012, 90, 201–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, P.J.; Lindert, U.; Opitz, L.; Hausser, I.; Rohrbach, M.; Giunta, C. Transcriptome Profiling of Primary Skin Fibroblasts Reveal Distinct Molecular Features Between PLOD1- and FKBP14-Kyphoscoliotic Ehlers-Danlos Syndrome. Genes 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, M.L.; Khattab, A. Genetics of congenital adrenal hyperplasia and genotype-phenotype correlation. Fertil. Steril. 2019, 111, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Hansen, U.; Keene, D.R.; Bruckner, P.; Chiquet-Ehrismann, R.; Chiquet, M.; Koch, M. Collagen XII interacts with avian tenascin-X through its NC3 domain. J. Biol. Chem. 2006, 281, 27461–27470. [Google Scholar] [CrossRef] [Green Version]
- Bristow, J.; Carey, W.; Egging, D.; Schalkwijk, J. Tenascin-X, Collagen, Elastin, and the Ehlers–Danlos Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2005, 139C, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbaere, S.; Dhooge, T.; Syx, D.; Petit, F.; Goemans, N.; Destrée, A.; Vanakker, O.; De Rycke, R.; Symoens, S.; Malfait, F. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet. Med. 2020, 22, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ith, B.; Wei, J.; Yet, S.F.; Perrella, M.A.; Layne, M.D. Aortic carboxypeptidase-like protein is expressed in collagen-rich tissues during mouse embryonic development. Gene Expr. Patterns 2005, 5, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Yet, S.F.; Maemura, K.; Hsieh, C.M.; Bernfield, M.; Perrella, M.A.; Lee, M.E. Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell. Biol. 2001, 21, 5256–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 28, 1853–1864. [Google Scholar] [CrossRef]
- Gagnon, A.; Landry, A.; Proulx, J.; Layne, M.D.; Sorisky, A. Aortic carboxypeptidase-like protein is regulated by transforming growth factor beta in 3T3-L1 preadipocytes. Exp. Cell Res. 2005, 308, 265–272. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Furuhashi, H.; Irie, R.; Nishikawa, M.; Yamamoto, J.; Hibi, T.; Miura, S.; Minamino, T.; et al. Aortic carboxypeptidase-like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 128, 1581–1596. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Couchman, J.R.; Pataki, C.A. An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 2012, 60, 885–897. [Google Scholar] [CrossRef]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113 Pt 2, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Aguirre-Negrete, M.G.; González-Flores, S.; Reynoso-Luna, M.C.; Fragoso, R.; Nazará, Z.; Tapia-Arizmendi, G.; Cantú, J.M. Ehlers-Danlos features with progeroid facies and mild mental retardation. Further delineation of the syndrome. Clin. Genet. 1986, 30, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Aguirre-Negrete, M.G.; Liparoli, J.C.; Cantú, J.M. Third case of a distinct variant of the Ehlers-Danlos Syndrome (EDS). Clin. Genet. 1981, 20, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Aguirre-Negrete, M.; Ramirez-Soltero, S.; González-Mendoza, A.; Martínez, R.M.; Velázquez-Cabrera, A.; Cantu, J. A distinct variant of the Ehlers-Danlos syndrome. Clin. Genet. 1979, 16, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Rosthøj, S.; Quentin, E.; Hollmann, J.; Glössl, J.; Okada, S.; Tønnesen, T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am. J. Hum. Genet. 1987, 41, 436–453. [Google Scholar] [PubMed]
- Quentin, E.; Gladen, A.; Rodén, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Fukumoto, S.; Furukawa, K.; Urano, T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [Green Version]
- Delbaere, S.; De Clercq, A.; Mizumoto, S.; Noborn, F.; Bek, J.W.; Alluyn, L.; Gistelinck, C.; Syx, D.; Salmon, P.L.; Coucke, P.J.; et al. b3galt6 Knock-Out Zebrafish Recapitulate β3GalT6-Deficiency Disorders in Human and Reveal a Trisaccharide Proteoglycan Linkage Region. Front. Cell Dev. Biol. 2020, 8, 597857. [Google Scholar] [CrossRef]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. A 2010, 152, 1333–1346. [Google Scholar] [CrossRef]
- Kosho, T.; Takahashi, J.; Ohashi, H.; Nishimura, G.; Kato, H.; Fukushima, Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A 2005, 138, 282–287. [Google Scholar] [CrossRef]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Lê, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Hatamochi, A.; Honda, T.; Yamaguchi, T.; Okamoto, N.; Miyake, N.; Yamada, S.; Sugahara, K. Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 2017, 50, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome--an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Walker, J.M.; Wang, F.; Park, J.G.; Palmer, A.E.; Giunta, C.; Rohrbach, M.; Steinmann, B.; Eide, D.J. Promotion of vesicular zinc efflux by ZIP13 and its implications for spondylocheiro dysplastic Ehlers-Danlos syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, E3530–E3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Abu, A.; Frydman, M.; Marek, D.; Pras, E.; Nir, U.; Reznik-Wolf, H.; Pras, E. Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am. J. Hum. Genet. 2008, 82, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Burkitt Wright, E.M.M.; Spencer, H.L.; Daly, S.B.; Manson, F.D.C.; Zeef, L.A.H.; Urquhart, J.; Zoppi, N.; Bonshek, R.; Tosounidis, I.; Mohan, M.; et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am. J. Hum. Genet. 2011, 88, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.K.; Gohil, K.; Packer, L.; Burk, R.F. Selenium deficiency, endurance exercise capacity, and antioxidant status in rats. J. Appl. Physiol. 1987, 63, 2532–2535. [Google Scholar] [CrossRef]
- Meani, N.; Pezzimenti, F.; Deflorian, G.; Mione, M.; Alcalay, M. The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS ONE 2009, 4, e4273. [Google Scholar] [CrossRef] [Green Version]
- Porter, L.F.; Gallego-Pinazo, R.; Keeling, C.L.; Kamieniorz, M.; Zoppi, N.; Colombi, M.; Giunta, C.; Bonshek, R.; Manson, F.D.; Black, G.C. Bruch’s membrane abnormalities in PRDM5-related brittle cornea syndrome. Orphanet J. Rare Dis. 2015, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbach, M.; Spencer, H.L.; Porter, L.F.; Burkitt-Wright, E.M.; Bürer, C.; Janecke, A.; Bakshi, M.; Sillence, D.; Al-Hussain, H.; Baumgartner, M.; et al. ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol. Genet. Metab. 2013, 109, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, R.; Kapferer-Seebacher, I.; Amberger, A.; Redolfi, R.; Dalonneau, F.; Björck, E.; Milnes, D.; Bally, I.; Rossi, V.; Thielens, N.; et al. C1R Mutations Trigger Constitutive Complement 1 Activation in Periodontal Ehlers-Danlos Syndrome. Front. Immunol. 2019, 10, 2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bork, P.; Beckmann, G. The CUB domain. A widespread module in developmentally regulated proteins. J. Mol. Biol. 1993, 231, 539–545. [Google Scholar] [CrossRef]
- Malfait, F.; Coucke, P.; Symoens, S.; Loeys, B.; Nuytinck, L.; De Paepe, A. The molecular basis of classic Ehlers-Danlos syndrome: A comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 2005, 25, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, M.; Vandersteen, A.; Yiş, U.; Serdaroglu, G.; Ataman, E.; Chopra, M.; Garcia, S.; Jones, K.; Kariminejad, A.; Kraenzlin, M.; et al. Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): Clinical, molecular and biochemical delineation. Orphanet J. Rare Dis. 2011, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Lautrup, C.K.; Teik, K.W.; Unzaki, A.; Mizumoto, S.; Syx, D.; Sin, H.H.; Nielsen, I.K.; Markholt, S.; Yamada, S.; Malfait, F.; et al. Delineation of musculocontractural Ehlers-Danlos Syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1197. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Watanabe, A.; Takeshita, H.; Matsumoto, K.I. A method for quantification of serum tenascin-X by nano-LC/MS/MS. Clin. Chim. Acta 2016, 459, 94–100. [Google Scholar] [CrossRef]
- Bruneel, A.; Dubail, J.; Roseau, C.; Prada, P.; Haouari, W.; Huber, C.; Dupré, T.; Poüs, C.; Cormier-Daire, V.; Seta, N. Serum bikunin is a biomarker of linkeropathies. Clin. Chim. Acta 2018, 485, 178–180. [Google Scholar] [CrossRef]
- Haouari, W.; Dubail, J.; Lounis-Ouaras, S.; Prada, P.; Bennani, R.; Roseau, C.; Huber, C.; Afenjar, A.; Colin, E.; Vuillaumier-Barrot, S.; et al. Serum bikunin isoforms in congenital disorders of glycosylation and linkeropathies. J. Inherit. Metab. Dis. 2020, 43, 1349–1359. [Google Scholar] [CrossRef]
- Vogel, A.; Holbrook, K.A.; Steinmann, B.; Gitzelmann, R.; Byers, P.H. Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab. Investig. 1979, 40, 201–206. [Google Scholar] [PubMed]





{kind=link}
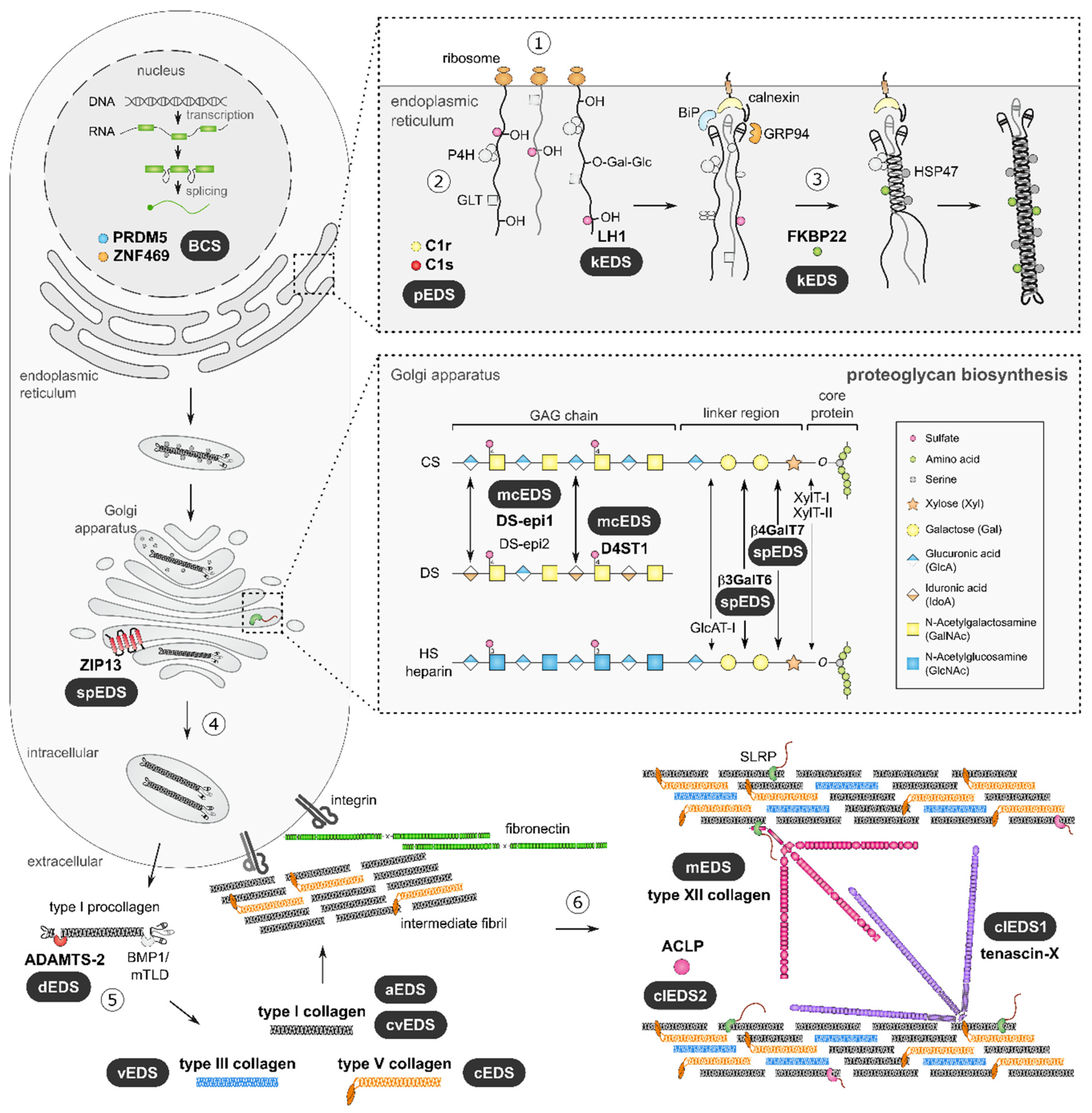{kind=link}
{kind=link}
{kind=link}
| EDS Type | Gene | Protein | IP | Estimated Incidence/Reported Individuals | Pathophysiological Mechanism | Biochemical Testing |
|---|---|---|---|---|---|---|
| Defects in Collagen Structure and Collagen Processing | ||||||
| Classical (cEDS) | COL5A1 COL5A2 (COL1A1 p.(Arg312Cys)) | α1-chain of type V procollagen α2-chain of type V procollagen α1-chain of type I procollagen | AD | 1:20,000 | Decreased type V collagen amounts affecting the initiation and assembly of heterotypic type I/V collagen fibrils Decreased type V collagen amounts affecting the initiation and assembly of heterotypic type I/V collagen fibrils Local destabilization of the type I collagen molecules and production of α1(I) dimers | (Pro)collagen biochemistry |
| Vascular (vEDS) | COL3A1 (COL1A1 p.(Arg312Cys), p.(Arg574Cys), p.(Arg1093Cys)) | α1-chain of type III procollagen α1-chain of type I procollagen | AD | 1:50,000–1:200,000 | Decreased type III collagen amounts affecting the initiation and assembly of heterotypic type I/III collagen fibrils Local destabilization of the type I collagen molecules and production of α1(I) dimers | (Pro)collagen biochemistry (Pro)collagen biochemistry |
| Arthrochalasia (aEDS) | COL1A1 COL1A2 | α1-chain of type I procollagen α2-chain of type I procollagen | AD | <60 reported individuals | (Partial or complete) deletion of exon 6 leading to partial processing of type I procollagen with retention of the N-propeptide of either the pro-α1(I)- or the pro-α2(I)-chain | (Pro)collagen biochemistry (Pro)collagen biochemistry |
| Dermatosparaxis (dEDS) | ADAMTS2 | A Disintegrin And Metalloproteinase with Thrombospondin Motifs 2 (ADAMTS-2) | AR | 15 reported individuals (14 families) | Absent N-propeptide cleavage of both pro-α(I)-chains | Procollagen biochemistry |
| Cardiac-valvular (cvEDS) | COL1A2 | α2-chain of type I procollagen | AR | 6 reported individuals (5 families) | Total absence of pro-α2(I)-chains leading to the formation of α1(I) homotrimers | (Pro)collagen biochemistry |
| Defects in Collagen Folding and Collagen Cross-Linking | ||||||
| Kyphoscoliotic (kEDS) | PLOD1 FKBP14 | Lysyl hydroxylase 1 (LH1) FK506 Binding Protein, 22 kDa (FKBP22) | AR | <100 reported individuals <35 reported individuals | Deficient post-translational hydroxylation of lysyl residues causing impaired crosslink formation in the collagen triple helix Deficiency of the molecular chaperone (possibly) leads to premature interaction and accumulation of collagen molecules in the endoplasmic reticulum | (Pro)collagen biochemistry Urinary crosslink analysis: increased LP/HP ratio (between 2–9) |
| Defects in Extracellular Matrix Bridging Molecules | ||||||
| Classical-like (clEDS) | TNXB | Tenascin X (TNX) | AR | <65 reported individuals | Interference with the normal organization and mechanical properties of collagen fibrils in the ECM | TNX serum levels |
| Myopathic (mEDS) | COL12A1 | α1-chain of type XII procollagen | AD AR | <20 reported individuals | Interference with the normal organization and mechanical properties of collagen fibrils in the ECM | NA |
| Defects in Intracellular Processes | ||||||
| Brittle cornea syndrome (BCS) | ZNF469 PRDM5 | Zinc Finger Protein 469 (ZNF469) PR/SET Domain 5 (PRDM5) | AR | <55 reported individuals <35 reported individuals | Disturbed ECM regulation, but exact pathophysiological mechanism remains unclear Disturbed ECM regulation, but exact pathophysiological mechanism remains unclear | NA NA |
| Spondylodysplastic (spEDS) | SLC39A13 | Zrt/Irt-Like Protein 13 (ZIP13) | AR | 13 reported individuals (7 families) | Generalized underhydroxylation of lysyl and prolyl residues of collagen and abnormal crosslinking of collagen in the ECM, but exact pathophysiological mechanism remains unclear | Urinary crosslink analysis: increased LP/HP ratio (around 1) |
| Defects in Glycosaminoglycan Biosynthesis | ||||||
| Musculocontractural (mcEDS) | CHST14 DSE | Dermatan 4-O-sulfotranferase-1 (D4ST1) Dermatan sulfate epimerase-1 (DS-epi1) | AR | <70 reported individuals | Defective biosynthesis of dermatan sulfate (DS) resulting in depletion of DS | Urinary disaccharide analysis Urinary disaccharide analysis |
| Spondylodysplastic (spEDS) | B4GALT7 B3GALT6 | Galactosyltransferase I (b4GalT7) Galactosyltransferase II (b3GalT6) | AR | <15 reported individuals <50 reported individuals | Absence of the first galactose residue of the tetrasacharide linker region of proteoglycans Absence of the second galactose residue of the tetrasacharide linker region of proteoglycans | Serum bikunin analysis Serum bikunin analysis |
| Defects in the Complement Pathway | ||||||
| Periodontal (pEDS) | C1S C1R | Complement C1s (C1s) Complement C1r (C1r) | AD | <150 reported individuals | Gain of function variants possibly leading to abnormal interactions with components of the ECM Gain of function variants possibly leading to abnormal interactions with components of the ECM | NA |
| Molecularly Unresolved | ||||||
| Hypermobile (hEDS) | ? | unknown | ? | ? | ? | NA |
| Novel Type of EDS (Identified After the 2017 Classification) | ||||||
| Classical-like II (clEDS II) | AEBP1 | Adipocyte enhancer-binding protein 1 (AEBP1) | AR | 9 reported individuals (9 families) | Interference with normal collagen fibril formation, but exact pathophysiological mechanism remains unclear | NA |
| EDS Type | Integumentary System | Skeletal System | Neuromuscular | Craniofacial | Ophthalmological | Vascular | Cardiac | Other |
|---|---|---|---|---|---|---|---|---|
| cEDS (COL5A1; COL5A2; COL1A1 p.(Arg312Cys)) | skin hyperextensibility with atrophic scarring, easy bruising soft doughy skin skin fragility, molluscoid pseudotumours, subcutaneous spheroids, hernia (or history thereof) | GJH, complications of JH | (mild) muscle hypotonia, delayed motor development | epicanthal folds | Rarely aortic root dilatation, rarely arterial dissection/rupture | MVP (non-progressive) | ||
| vEDS (COL3A1; COL1A1 p.(Arg312Cys, p.(Arg574Cys), p.(Arg1093Cys) | bruising unrelated to identified trauma and/or in unusual sites, translucent skin, acrogeria | talipes equinovarus, congenital hip dislocation, small joint hypermobility, tendon and muscle rupture | characteristic facial features (large eyes, periorbital pigmentation, small chin, sunken cheeks, thin nose and lips and lobeless ears), gingival recession and gingival fragility | keratoconus | arterial rupture at young age, carotid-cavernous sinus fistula, early-onset varicose veins | spontaneous sigmoid colon perforation, uterine rupture during third trimester of pregnancy, spontaneous pneumothorax | ||
| aEDS (COL1A1; COL1A2) | skin hyperextensibility, tissue fragility including atrophic scars, easy bruising, umbilical hernia | congenital bilateral hip dislocation, severe generalized JH with multiple dislocations, kyphoscoliosis, radiologically mild osteopenia, fractures, foot and hand deformities, pectus deformity | muscle hypotonia, delayed motor development | large fontanelle, frontal bossing, hypertelorism, blue sclerae, epicanthal folds, depressed nasal ridge, midface hypoplasia, micrognathia, dentinogenesis imperfecta | pregnancy-related complications (breech, PPROM, polyhydramnios, decreased fetal movements) | |||
| dEDS (ADAMTS2) | extreme skin fragility with congenital or postnatal tears, progressively redundant, almost lax skin with excessive skin folds at wrists and ankles, increased palmar wrinkling, severe bruisability with risk of subcutaneous haematoma, umbilical hernia, soft and doughy skin texture, skin hyperextensibility, atrophic scars, hirsutism | postnatal growth retardation with short limbs, GJH, osteopenia, fractures | delayed motor development | large fontanel, puffy eyelids, excessive peri-orbital skin, downslanting palpebral fissures, blue sclerae, hypoplastic chin, tooth abnormalities | refractive errors, strabismus, glaucoma | Intracerebral hemorrhage | perinatal complications related to tissue fragility, complications of visceral fragility, preterm birth (PPROM), | |
| cvEDS (COL1A1; COL1A2) | skin involvement, inguinal hernia | JH, pectus deformity, joint dislocations, foot deformities | blue sclerae, refractive errors | severe progressive cardiac valvular insufficiency | ||||
| kEDS (PLOD1; FKBP14) | skin hyperextensibility, easy bruising, umbilical or inguinal hernia PLOD1: skin fragility FKBP14: hyperkeratosis follicularis, umbilical skin redundancy | congenital or early-onset kyphoscoliosis, GJH with (sub)luxations, osteopenia/osteoporosis, pectus deformity, marfanoid habitus, talipes equinovarus | congenital muscle hypotonia, delayed motor development FKBP14: muscle atrophy | blue sclerae PLOD1: characteristic craniofacial features: low-set ears, epicanthal folds, down-slanting palpebral fissures, synophrys and high palate | refractive errors PLOD1: microcornea | rupture/aneurysm of medium-sized artery, antenatal/neonatal brain hemorrhage | pregnancy-related complications (breech, PPROM, oligohydramnios, decreased fetal movements) FKBP14: congenital hearing impairment, bladder diverticula, learning disabilities | |
| clEDS-1 (TNXB) | skin hyperextensibility with velvety skin texture and absence of (extensive) atrophic scarring, easily bruisable skin/spontaneous ecchymoses, acrogeric hands | GJH, foot deformities, mallet fingers, clino- or brachydactyly, (sub)luxations | mild proximal and distal muscle weakness, axonal polyneuropathy, atrophy of muscle in hands and feet | narrow/high arched palate | rarely arterial aneurysms | valvular abnormalities | oedema in legs in absence of cardiac failure, vaginal, uterine or rectal prolapse, gastrointestinal complications (perforation, diverticular disease, …), postpartum hemorrhage | |
| clEDS2 (AEBP1) | skin hyperextensibility with atrophic scarring, translucent skin, easy bruising, hernia, aged appearance | GJH, foot deformities, early-onset osteopenia, joint dislocations (mostly hip), arachnodactyly, (kypho)scoliosis, osteopenia | bad tooth quality, dental abnormalities, high narrow palate, | varicose veins, aorta dilatation | MVP | bowel rupture | ||
| mEDS (COL12A1) | soft, doughy skin, atrophic scars, hypertrophic scars | proximal joint contractures, (general/distal) JH, congenital hip dislocation, congenital (kypho)scoliosis, pectus deformity | congenital muscle hypotonia and/or muscle atrophy, motor developmental delay, myopathy on muscle biopsy | |||||
| BCS (ZNF469; PRDM5) | soft, velvety and/or translucent skin, mild skin hyperextensibility, easy bruising, hernia | developmental dysplasia of hip, scoliosis, arachnodactyly, hypermobility of distal joints, pes planus, hallux valgus, mild finger contractures, fractures, osteopenia/osteoporosis | hypotonia in infancy (usually mild) | blue sclerae, frontal bossing, high palate, depressed nasal bridge and/or prominent chin | thin cornea with/without rupture, early-onset progressive keratoconus and/or keratoglobus, enucleation or corneal scarring because of previous rupture, progressive loss of corneal stromal depth, high myopia, retinal detachment | MVP (non-progressive) | deafness (often mixed conductive and sensorineural), hypercompliant tympanic membranes | |
| mcEDS (CHST14; DSE) | skin hyperextensibility, easy bruising, skin fragility with atrophic scars, increased palmar wrinkling, large subcutaneous hematomas, hernia | Recurrent/chronic dislocations, pectus deformities, spinal deformities, peculiar fingers, progressive talipes deformities, mild postnatal growth restriction, marfanoid habitus | congenital multiple contractures (typically adduction/flexion contractures and talipes equinovarus), hypotonia, motor developmental delay, ventricular abnormalities on brain imaging, tethered spinal cord | large fontanelle, short downslanting palpebral fissures, blue sclerae, hypertelorism, short nose with hypoplastic columella, low-set and rotated ears, long philtrum with thin upper lip vermillion, small mouth and hypoplastic chin, crowded teeth | strabismus, refractive errors, glaucoma, retinal detachment | congenital heart defects (typically ASD), valve abnormalities, aortic root dilatation | chronic constipation, colonic diverticulae, pneumo(haemo)thorax, nephrolithiasis/cystolithiasis, hydronephrosis, cryptorchidism in males, hearing impairment, constipation, diverticula, poor breast development in females | |
| spEDS (B3GALT6; B4GALT7; SLC39A13) | skin hyperextensibility, soft and doughy, thin and translucent skin B4GALT7: single transverse palmar crease SLC39A13: hands with finely wrinkled palms | short stature (progressive in childhood), bowing of limbs, pes planus, osteopenia, (characteristic X-ray findings of) skeletal dysplasia, JH B4GALT7: radioulnar synostosis B3GALT6: kyphoscoliosis (congenital or early-onset), JH (generalized or restricted to distal joints), peculiar fingers, osteoporosis with spontaneous fractures SLC39A13: tapering fingers, hypermobility of distal joints | muscle hypotonia (ranging from severe congenital to mild later-onset), delayed motor development B4GALT7: bilateral elbow contractures B3GALT6: joint contractures (congenital or progressive) SLC39A13: atrophy of thenar muscles | B4GALT7: triangular face, wide-spaced eyes, proptosis, narrow mouth, low-set ears, sparse scalp hair, abnormal dentition, flat face, wide forehead, blue sclerae and cleft palate/bifid uvula B3GALT6: midfacial hypoplasia, frontal bossing, proptosis, or prominent eyes, blue sclerae, downslanting palpebral fissures, depressed nasal bridge, long upper lip, low-set ears, micrognathia, abnormal dentition, cleft palate, sparse hair, tooth discoloration, dysplastic teeth SLC39A13: protuberant eyes with bluish sclerae, hypodontia of one or few teeth | B4GALT7: clouded cornea, refractive errors B3GALT6: hypermetropia, rarely corneal clouding SLC39A13: refractive errors | Rarely aortic aneurysm SLC39A13: varicose veins | B3GALT6: MVP, congenital heart defects | cognitive impairment B3GALT6: lung hypoplasia, restrictive lung disease |
| pEDS (C1R; C1S) | pretibial plaques, easy bruising, skin hyperextensibility and fragility, wide or atrophic scarring, hernias, acrogeria, skin translucency | JH (mostly distal), kypho (scoliosis) | Severe and intractable early-onset periodontitis, lack of attached gingiva, marfanoid facial features | prominent vasculature, rarely arterial dissection/rupture | family history of first-degree relative who meets clinical criteria, increased infection rate | |||
| hEDS (genetic defect unknown) | unusually soft or velvety skin, mild skin hyperextensibility, unexplained striae, bilateral piezogenic papules, hernia, mild atrophic scarring, | GJH, arachnodactyly, arm span to height ratio ≥1.05, | dental crowding and high or narrow palate | aortic root dilatation with z score > +2 | MVP | pelvic floor, rectal and/or uterine prolapse, positive family history of hEDS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Damme, T.; Colman, M.; Syx, D.; Malfait, F. The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes 2022, 13, 265. https://doi.org/10.3390/genes13020265
Van Damme T, Colman M, Syx D, Malfait F. The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes. 2022; 13(2):265. https://doi.org/10.3390/genes13020265
Chicago/Turabian StyleVan Damme, Tim, Marlies Colman, Delfien Syx, and Fransiska Malfait. 2022. "The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism" Genes 13, no. 2: 265. https://doi.org/10.3390/genes13020265
APA StyleVan Damme, T., Colman, M., Syx, D., & Malfait, F. (2022). The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes, 13(2), 265. https://doi.org/10.3390/genes13020265