
Clinical and Genetic Aspects of Phelan–McDermid Syndrome: An Interdisciplinary Approach to Management



 , and
, and
Abstract
:1. Introduction
2. Clinical Features
Natural History
3. Etiopathogenesis
4. Molecular Diagnosis
5. Differential Clinical Diagnosis
6. Interdisciplinary Clinical Management
7. Therapeutic Options
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kniffin, C.L.; McKusick, V.A. #606232 Phelan–McDermid Syndrome, PHMDS. Online Mendelian Inheritance in Man, OMIM®. 2015. Available online: https://www.omim.org/entry/606232 (accessed on 12 December 2021).
- Boccuto, L.; Abenavoli, L.; Cascio, L.; Srikanth, S.; Dupont, B.; Mitz, A.R.; Rogers, R.C.; Phelan, K. Variability in Phelan-McDermid syndrome: The impact of the PNPLA3 p.I148M polymorphism. Clin. Genet. 2018, 94, 590–591. [Google Scholar] [CrossRef]
- Drapeau, E.; Riad, M.; Kajiwara, Y.; Buxbaum, J.D. Behavioral Phenotyping of an Improved Mouse Model of Phelan–McDermid Syndrome with a Complete Deletion of the Shank3 Gene. eNeuro 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtas, N.; Arrigoni, F.; Errichiello, E.; Zucca, C.; Maghini, C.; D’Angelo, M.G.; Beri, S.; Giorda, R.; Bertuzzo, S.; Delledonne, M.; et al. Chromothripsis and ring chromosome 22: A paradigm of genomic complexity in the Phelan–McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2018, 55, 269–277. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.D.; Trana, C.J.; Kelsay, J.; Sawyer, J.; Clothier, J. Phelan–McDermid syndrome and cancer predisposition: The value of a karyotype. Am. J. Med. Genet. Part A 2018, 176, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Mitz, A.R.; Philyaw, T.J.; Boccuto, L.; Shcheglovitov, A.; Sarasua, S.M.; Kaufmann, W.E.; Thurm, A. Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur. J. Hum. Genet. 2018, 26, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Ziats, C.A.; Grosvenor, L.P.; Sarasua, S.M.; Thurm, A.E.; Swedo, S.E.; Mahfouz, A.; Rennert, O.M.; Ziats, M.N. Functional genomics analysis of Phelan–McDermid syndrome 22q13 region during human neurodevelopment. PLoS ONE 2019, 14, e0213921. [Google Scholar] [CrossRef] [Green Version]
- Zwanenburg, R.J.; Bocca, G.; Ruiter, S.A.J.; Dillingh, J.H.; Flapper, B.C.T.; Heuvel, E.R.V.D.; Van Ravenswaaij-Arts, C.M.A. Is there an effect of intranasal insulin on development and behaviour in Phelan–McDermid syndrome? A randomized, double-blind, placebo-controlled trial. Eur. J. Hum. Genet. 2016, 24, 1696–1701. [Google Scholar] [CrossRef] [Green Version]
- Phelan, K.; McDermid, H. The 22q13.3 Deletion Syndrome (Phelan–McDermid Syndrome). Mol. Syndr. 2011, 2, 186–201. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.; Powis, L.; Moss, J.; Stinton, C.; Nelson, L.; Oliver, C. Prospective study of autism phenomenology and the behavioural phenotype of Phelan–McDermid syndrome: Comparison to fragile X syndrome, Down syndrome and idiopathic autism spectrum disorder. J. Neurodev. Disord. 2017, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.L.; Wong, A.C.C.; Shaw, S.R.; Tse, W.-Y.; Stapleton, G.A.; Phelan, M.C.; Hu, S.; Marshall, J.; McDermid, H.E. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 2003, 40, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Costales, J.L.; Kolevzon, A. Phelan–McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics 2015, 12, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disciglio, V.; Rizzo, C.L.; Mencarelli, M.A.; Mucciolo, M.; Marozza, A.; Di Marco, C.; Massarelli, A.; Canocchi, V.; Baldassarri, M.; Ndoni, E.; et al. Interstitial 22q13 deletions not involving SHANK3 gene: A new contiguous gene syndrome. Am. J. Med. Genet. Part A 2014, 164, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Tabet, A.-C.; Rolland, T.; Ducloy, M.; Levy, J.; Buratti, J.; Mathieu, A.; Haye, D.; Perrin, L.; Dupont, C.; Passemard, S.; et al. A framework to identify contributing genes in patients with Phelan–McDermid syndrome. NPJ Genom. Med. 2017, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Borgatti, R.; Felisari, G.; Gagliardi, C.; Selicorni, A.; Zuffardi, O. Disruption of the ProSAP2 Gene in a t(12;22)(q24.1;q13.3) Is Associated with the 22q13.3 Deletion Syndrome. Am. J. Hum. Genet. 2001, 69, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Watt, J.L.; Olson, I.A.; Johnston, A.W.; Ross, H.S.; Couzin, D.A.; Stephen, G.S. A familial pericentric inversion of chromosome 22 with a recombinant subject illustrating a ‘pure’ partial monosomy syndrome. J. Med. Genet. 1985, 22, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Herman, G.E.; Greenberg, F.; Ledbetter, D.H.; Optiz, J.M.; Reynolds, J.F. Multiple congenital anomaly/mental retardation (MCA/MR) syndrome with Goldenhar complex due to a terminal del(22q). Am. J. Med. Genet. 1988, 29, 909–915. [Google Scholar] [CrossRef]
- Phelan, M.C.; Rogers, R.C.; Stevenson, R.E. A de Novo Terminal Deletion of 22q. Am. J. Hum. Genet. 1988, 43, A118. [Google Scholar]
- Egger, J.I.M.; Zwanenburg, R.J.; Ravenswaaij-Arts, C.M.A.; Kleefstra, T.; Verhoeven, W.M.A. Neuropsychological phenotype and psychopathology in seven adult patients with Phelan–McDermid syndrome: Implications for treatment strategy. Genes Brain Behav. 2016, 15, 395–404. [Google Scholar] [CrossRef]
- Reierson, G.; Bernstein, J.; Froehlich-Santino, W.; Urban, A.; Purmann, C.; Berquist, S.; Jordan, J.; O’Hara, R.; Hallmayer, J. Characterizing regression in Phelan McDermid Syndrome (22q13 deletion syndrome). J. Psychiatr. Res. 2017, 91, 139–144. [Google Scholar] [CrossRef]
- Tabolacci, E.; Zollino, M.; Lecce, R.; Sangiorgi, E.; Gurrieri, F.; Leuzzi, V.; Opitz, J.M.; Neri, G. Two Brothers with 22q13 Deletion Syndrome and Features Suggestive of the Clark–Baraitser Syndrome. Clin. Dysmorphol. 2005, 14, 127–132. [Google Scholar] [CrossRef]
- Philippe, A.; Boddaert, N.; Vaivre-Douret, L.; Robel, L.; Danon-Boileau, L.; Malan, V.; de Blois, M.-C.; Heron, D.; Colleaux, L.; Golse, B.; et al. Neurobehavioral Profile and Brain Imaging Study of the 22q13.3 Deletion Syndrome in Childhood. Pediatrics 2008, 122, e376–e382. [Google Scholar] [CrossRef]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of Phelan–McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, H.L.; Crolla, J.A.; Walker, D.; Artifoni, L.; Dallapiccola, B.; Takano, T.; Vasudevan, P.; Huang, S.; Maloney, V.; Yobb, T.; et al. Interstitial 22q13 deletions: Genes other than SHANK3 have major effects on cognitive and language development. Eur. J. Hum. Genet. 2008, 16, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Culotta, L.; Scalmani, P.; Vinci, E.; Terragni, B.; Sessa, A.; Broccoli, V.; Mantegazza, M.; Verpelli, C. SULT4A1 modulates synaptic development and function by promoting the formation of PSD-95/NMDAR complex. J. Neurosci. 2020, 40, 7013–7026. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2006, 39, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Nemirovsky, S.I.; Córdoba, M.; Zaiat, J.J.; Completa, S.P.; Vega, P.A.; González-Morón, D.; Medina, N.M.; Fabbro, M.; Romero, S.; Brun, B.; et al. Whole Genome Sequencing Reveals a De Novo SHANK3 Mutation in Familial Autism Spectrum Disorder. PLoS ONE 2015, 10, e0116358. [Google Scholar] [CrossRef]
- Zirn, B.; Arning, L.; Bartels, I.; Shoukier, M.; Hoffjan, S.; Neubauer, B.; Hahn, A. Ring chromosome 22 and neurofibromatosis type II: Proof of two-hit model for the loss of the NF2 gene in the development of meningioma. Clin. Genet. 2010, 81, 82–87. [Google Scholar] [CrossRef]
- Ziats, C.A.; Jain, L.; McLarney, B.; Vandenboom, E.; DuPont, B.R.; Rogers, C.; Sarasua, S.; Nevado, J.; Cordisco, E.L.; Phelan, K.; et al. Neurofibromatosis type 2 in Phelan–McDermid syndrome: Institutional experience and review of the literature. Eur. J. Med Genet. 2020, 63, 104042. [Google Scholar] [CrossRef]
- Lyons-Warren, A.M.; Cheung, S.W.; Lloyd Holder, J., Jr. Clinical Reasoning: A common cause for Phelan–McDermid syndrome and neurofibromatosis type 2. Neurology 2017, 89, e205–e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woike, D.; Wang, E.; Tibbe, D.; Nia, F.H.; Failla, A.V.; Kibæk, M.; Overgård, T.M.; Larsen, M.J.; Fagerberg, C.R.; Barsukov, I.; et al. Mutations affecting the N-terminal domains of SHANK3 point to different pathomechanisms in neurodevelopmental disorders. Sci. Rep. 2022, 12, 902. [Google Scholar] [CrossRef] [PubMed]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.-F.; Rogers, R.C.; Phelan, K.; DuPont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan–McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.-L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021, 26, 7596–7609. [Google Scholar] [CrossRef]
- Ma, K.; Qin, L.; Matas, E.; Duffney, L.J.; Liu, A.; Yan, Z. Histone deacetylase inhibitor MS-275 restores social and synaptic function in a Shank3-deficient mouse model of autism. Neuropsychopharmacology 2018, 43, 1779–1788. [Google Scholar] [CrossRef]
- Breen, M.S.; Browne, A.; Hoffman, G.E.; Stathopoulos, S.; Brennand, K.; Buxbaum, J.D.; Drapeau, E. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan–McDermid syndrome and autism. Mol. Autism 2020, 11, 53. [Google Scholar] [CrossRef]
- Lin, R.; Learman, L.N.; Bangash, M.A.; Melnikova, T.; Leyder, E.; Reddy, S.C.; Naidoo, N.; Park, J.M.; Savonenko, A.; Worley, P.F. Homer1a regulates Shank3 expression and underlies behavioral vulnerability to stress in a model of Phelan–McDermid syndrome. Cell Rep. 2021, 37, 110014. [Google Scholar] [CrossRef]
- Zwanenburg, R.J.; Ruiter, S.A.; Heuvel, E.R.V.D.; Flapper, B.C.; Van Ravenswaaij-Arts, C.M. Developmental phenotype in Phelan–McDermid (22q13.3 deletion) syndrome: A systematic and prospective study in 34 children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Harony-Nicolas, H.; De Rubeis, S.; Kolevzon, A.; Buxbaum, J.D. Phelan McDermid Syndrome. J. Child Neurol. 2015, 30, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Kolevzon, A.; Angarita, B.; Bush, L.; Wang, A.T.; Frank, Y.; Yang, A.; Rapaport, R.; Saland, J.; Srivastava, S.; Farrell, C.; et al. Phelan–McDermid syndrome: A review of the literature and practice parameters for medical assessment and monitoring. J. Neurodev. Disord. 2014, 6, 39. [Google Scholar] [CrossRef]
- Omansky, G.L.; Abdulhayoglu, E.; Zhurbilo, B.; Leary, O.G.; Elisa, A.; Bella, Z. Phelan–McDermid Syndrome. Neonatal Netw. 2017, 36, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Kern, W.; Giese, R.; Hallschmid, M.; Enders, A. Intranasal insulin to improve developmental delay in children with 22q13 deletion syndrome: An exploratory clinical trial. J. Med. Genet. 2008, 46, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Rajamani, K.T.; Wagner, S.; Grinevich, V.; Harony-Nicolas, H. Oxytocin as a Modulator of Synaptic Plasticity: Implications for Neurodevelopmental Disorders. Front. Synaptic Neurosci. 2018, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Verpelli, C.; Dvoretskova, E.; Vicidomini, C.; Rossi, F.; Chiappalone, M.; Schoen, M.; Di Stefano, B.; Mantegazza, R.; Broccoli, V.; Böckers, T.M.; et al. Importance of Shank3 Protein in Regulating Metabotropic Glutamate Receptor 5 (mGluR5) Expression and Signaling at Synapses. J. Biol. Chem. 2011, 286, 34839–34850. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.; Reim, D.; Morello, N.; Orelanna, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2016, 22, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Serret, S.; Thümmler, S.; Dor, E.; Vesperini, S.; Santos, A.; Askenazy, F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: Case reports. BMC Psychiatry 2015, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Dyar, B.; Meaddough, E.; Sarasua, S.; Rogers, C.; Phelan, K.; Boccuto, L. Genetic Findings as the Potential Basis of Personalized Pharmacotherapy in Phelan–McDermid Syndrome. Genes 2021, 12, 1192. [Google Scholar] [CrossRef]
- Qin, L.; Ma, K.; Wang, Z.-J.; Hu, Z.; Matas, E.; Wei, J.; Yan, Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 2018, 21, 564–575. [Google Scholar] [CrossRef]
- Qin, L.; Ma, K.; Yan, Z. Rescue of histone hypoacetylation and social deficits by ketogenic diet in a Shank3 mouse model of autism. Neuropsychopharmacology 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Torrioli, M.; Vernacotola, S.; Setini, C.; Bevilacqua, F.; Martinelli, D.; Snape, M.; Hutchison, J.A.; Di Raimo, F.R.; Tabolacci, E.; Neri, G. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am. J. Med. Genet. Part A 2010, 152A, 1420–1427. [Google Scholar] [CrossRef] [PubMed]


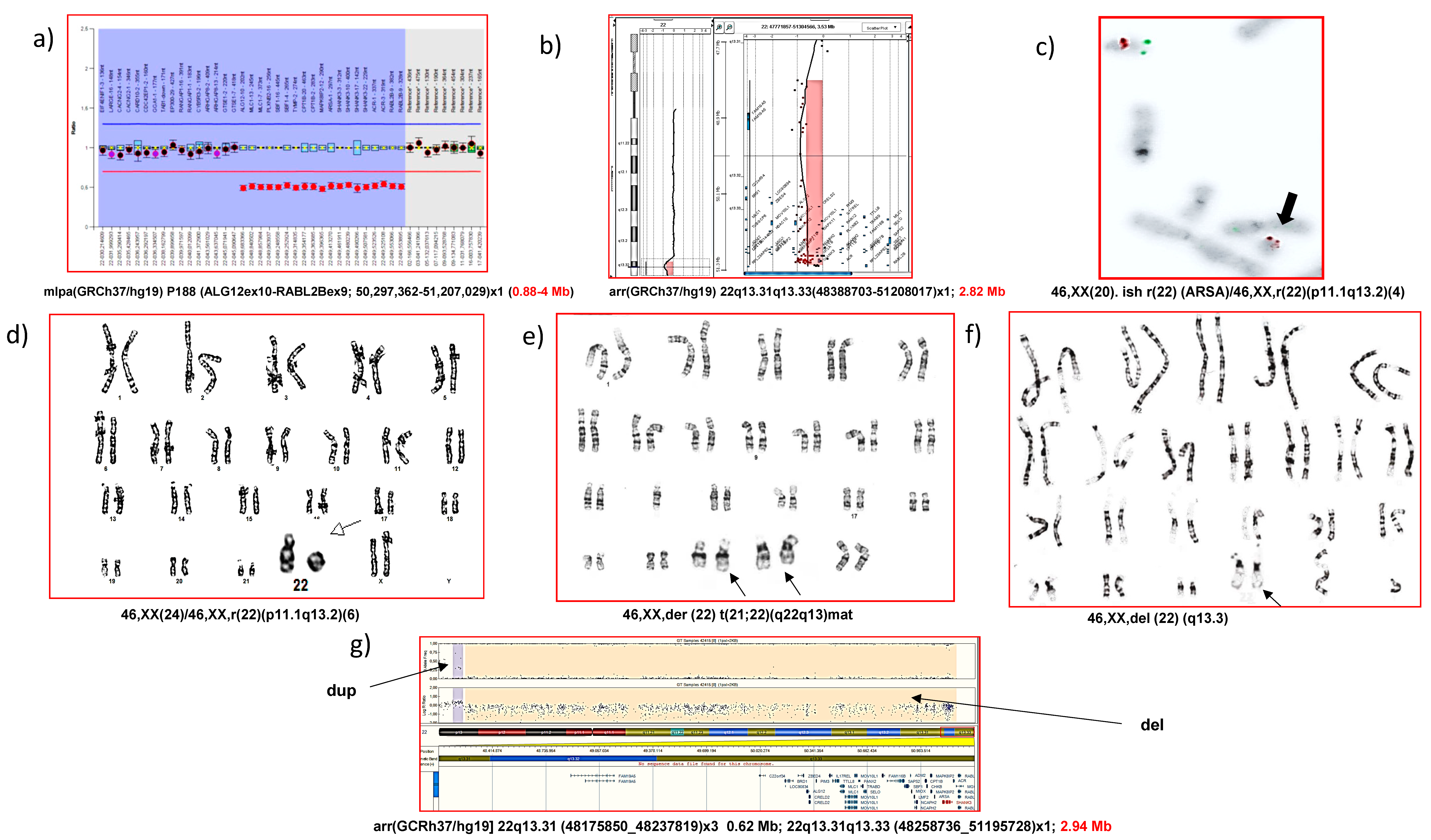{kind=link}
| Findings | Percentage (%) | Findings | Percentage (%) |
|---|---|---|---|
| Hyperextensibility | 86 | Dimple in sacrum | 25 |
| Hypotonia | 73 | Lymphedema | 25 |
| Bulbous nasal tip | 65 | Febrile/nonfebrile seizure | 24 |
| Alterations in ears | 63 | Hypertelorism | 22 |
| Long eyelashes | 58 | Kyphoscoliosis | 22 |
| Large, fleshy hands | 53 | Sunken eyes | 19 |
| Epicanthal folds | 48 | Malocclusion/separated teeth | 19 |
| Periorbital fullness | 46 | Macrocephaly | 17 |
| Dysplastic/hypoplastic nails | 45 | Esotropia/strabismus | 17 |
| Pointed chin | 44 | Sparse/spiral hair | 16 |
| Sleep disturbance | 44 | Wide nasal bridge | 16 |
| Gastroesophageal reflux | 43 | Long philtrum | 16 |
| Increased tolerance to pain | 42 | Microcephaly | 12 |
| Constipation/diarrhea | 40 | Micrognathia | 12 |
| Dolichocephaly | 37 | clinodactyly of the fifth finger | 12 |
| High narrow palate | 36 | Short stature/growth retardation | 12 |
| Syndactyly of 2nd and 3rd toes | 34 | Tall stature/accelerated growth | 11 |
| Brain abnormalities (imaging) | 32 | Malar hypoplasia | 9 |
| Prominent lips | 31 | Congenital heart disease | 8 |
| Recurrent respiratory infections | 30 | Precocious or delayed puberty | 6 |
| Eyelid ptosis | 29 | Low-set ears | 5 |
| Kidney disorders | 27 | Hypothyroidism | 5 |
| Prominent cheeks | 25 | Midface hypoplasia | 3 |
| Large Deletions | Cryptic Deletions | Balanced Translocations | Size | Mosaics | UPD | Other Regions | |
|---|---|---|---|---|---|---|---|
| Karyotype | + | − | + | − | + | + | + Large |
| FISH | + | + | + | − | + | + | − |
| MLPA | + | + | − | + Partial | − | + | − |
| aCGH | + | + | − Yes, in imbalance | + | + | + | + |
| SNParray | + | + | − Yes, in imbalance | + | + | − | + |
| Findings/Entities | PMS | Autism | Paralysis Cerebral | Prader- Willi | Angelman | Velocardiofacial | Fragile X | FG | Williams | Smith- Magenis |
|---|---|---|---|---|---|---|---|---|---|---|
| Subtle dysmorphisms | + | − | + | + | + | + | + | − * | + ** | + |
| Language disturbance | + | + | + | + | + | + | + | − | + | |
| Alteration in socialization | + | + | − | − | + | − | + | − | − | − |
| Repetitive movements | + | + | − | − | + | − | + | + | − | − |
| Hypotonia | + | − | + | + | + | + | − | + | + | + |
| Global developmental delay | + | − | + | + | + | + | + | + | + | + |
| Feeding difficulties | + | − | + | + | − | + | − | − | + | − |
| Poor coordination | + | − | + | − | + | − | + | − | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammarata-Scalisi, F.; Callea, M.; Martinelli, D.; Willoughby, C.E.; Tadich, A.C.; Araya Castillo, M.; Lacruz-Rengel, M.A.; Medina, M.; Grimaldi, P.; Bertini, E.; et al. Clinical and Genetic Aspects of Phelan–McDermid Syndrome: An Interdisciplinary Approach to Management. Genes 2022, 13, 504. https://doi.org/10.3390/genes13030504
Cammarata-Scalisi F, Callea M, Martinelli D, Willoughby CE, Tadich AC, Araya Castillo M, Lacruz-Rengel MA, Medina M, Grimaldi P, Bertini E, et al. Clinical and Genetic Aspects of Phelan–McDermid Syndrome: An Interdisciplinary Approach to Management. Genes. 2022; 13(3):504. https://doi.org/10.3390/genes13030504
Chicago/Turabian StyleCammarata-Scalisi, Francisco, Michele Callea, Diego Martinelli, Colin Eric Willoughby, Antonio Cárdenas Tadich, Maykol Araya Castillo, María Angelina Lacruz-Rengel, Marco Medina, Piercesare Grimaldi, Enrico Bertini, and et al. 2022. "Clinical and Genetic Aspects of Phelan–McDermid Syndrome: An Interdisciplinary Approach to Management" Genes 13, no. 3: 504. https://doi.org/10.3390/genes13030504
APA StyleCammarata-Scalisi, F., Callea, M., Martinelli, D., Willoughby, C. E., Tadich, A. C., Araya Castillo, M., Lacruz-Rengel, M. A., Medina, M., Grimaldi, P., Bertini, E., & Nevado, J. (2022). Clinical and Genetic Aspects of Phelan–McDermid Syndrome: An Interdisciplinary Approach to Management. Genes, 13(3), 504. https://doi.org/10.3390/genes13030504