
Inherited and De Novo Variation in Lithuanian Genomes: Introduction to the Analysis of the Generational Shift

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Sequencing
2.3. Structural Variation Detection
2.4. De Novo Mutation Detection
2.5. Variant Analysis
2.6. Variant Annotation
3. Results
3.1. Sample Collection
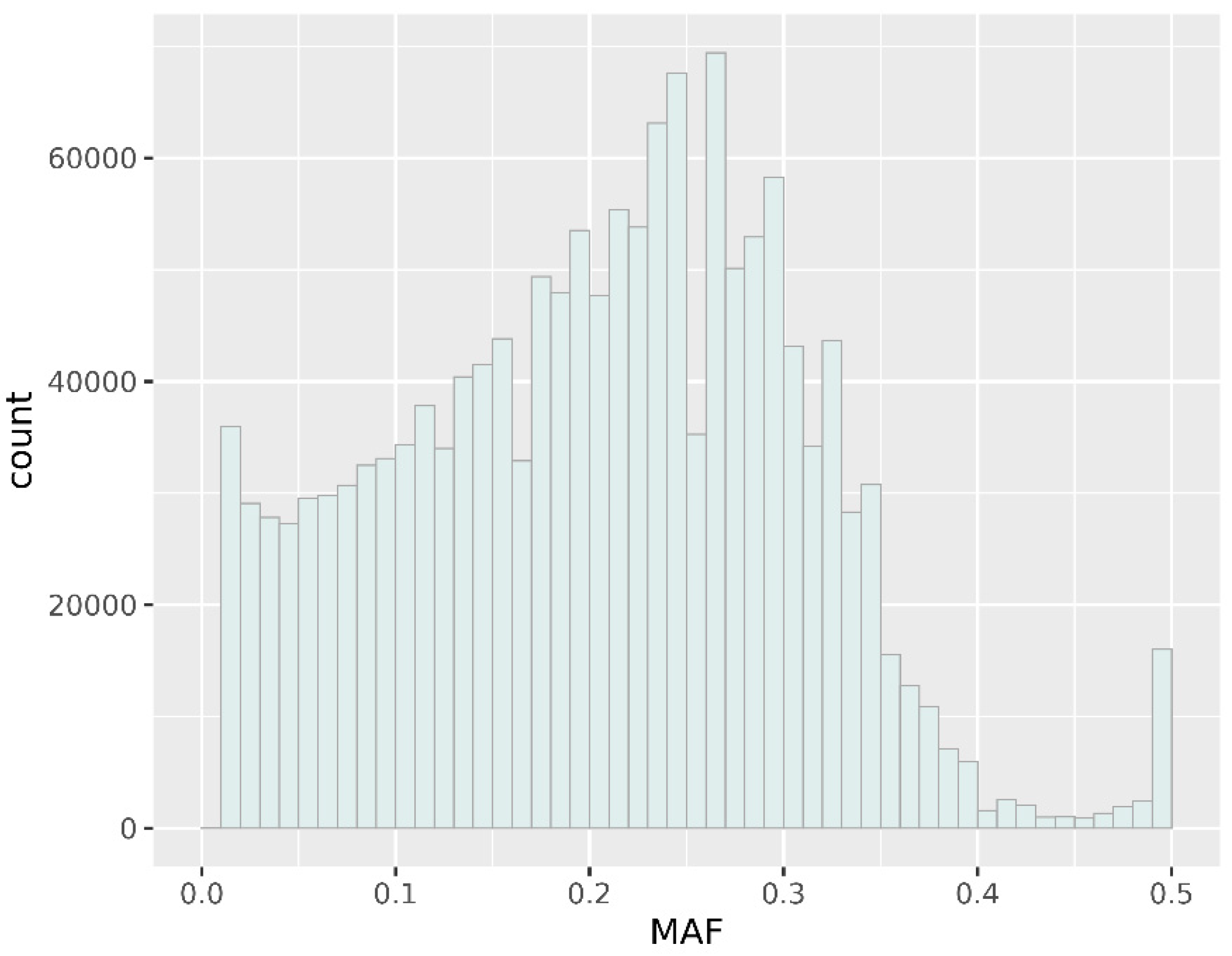
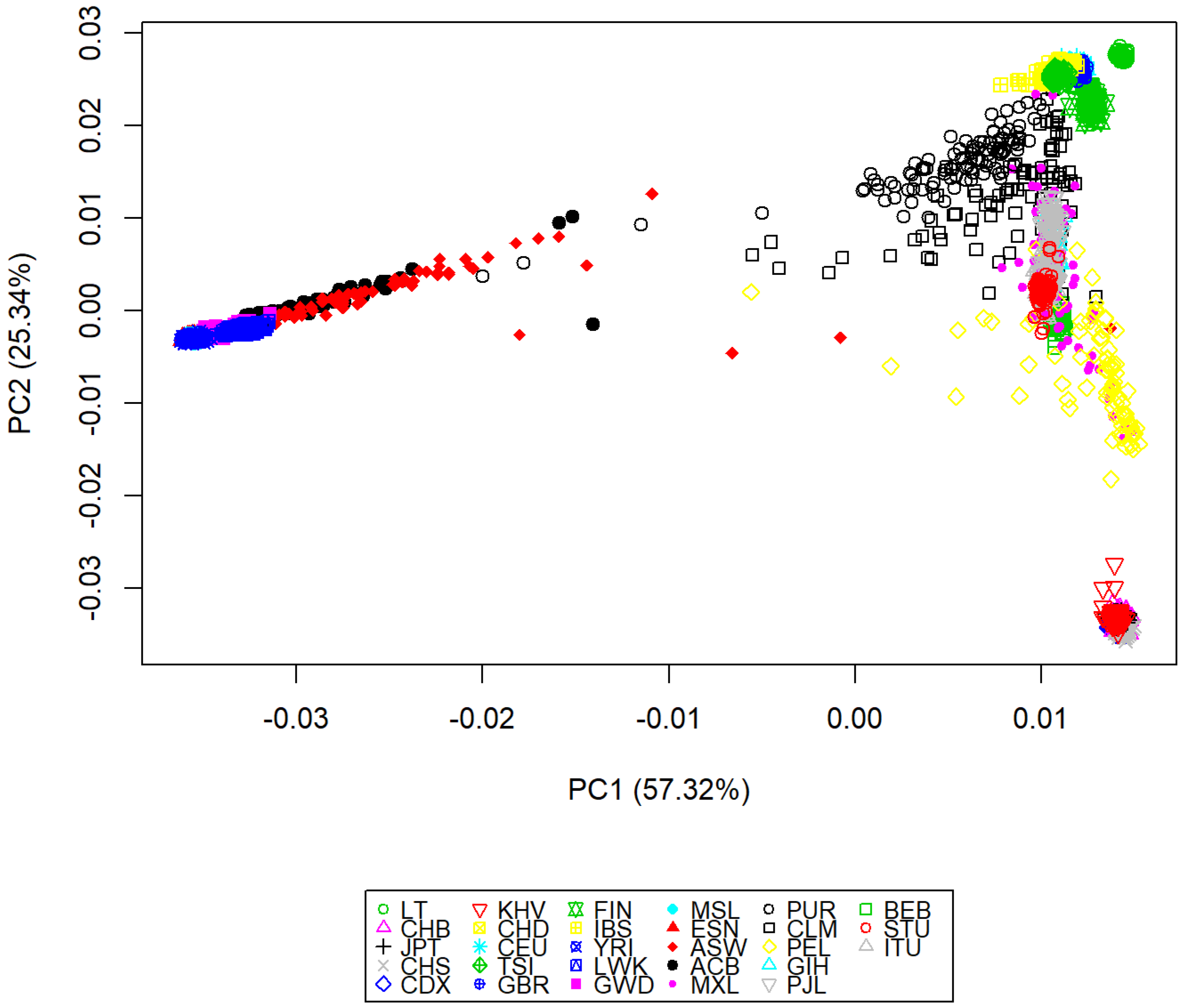
3.2. Genomic Variation Characterization
3.3. Structural Variation
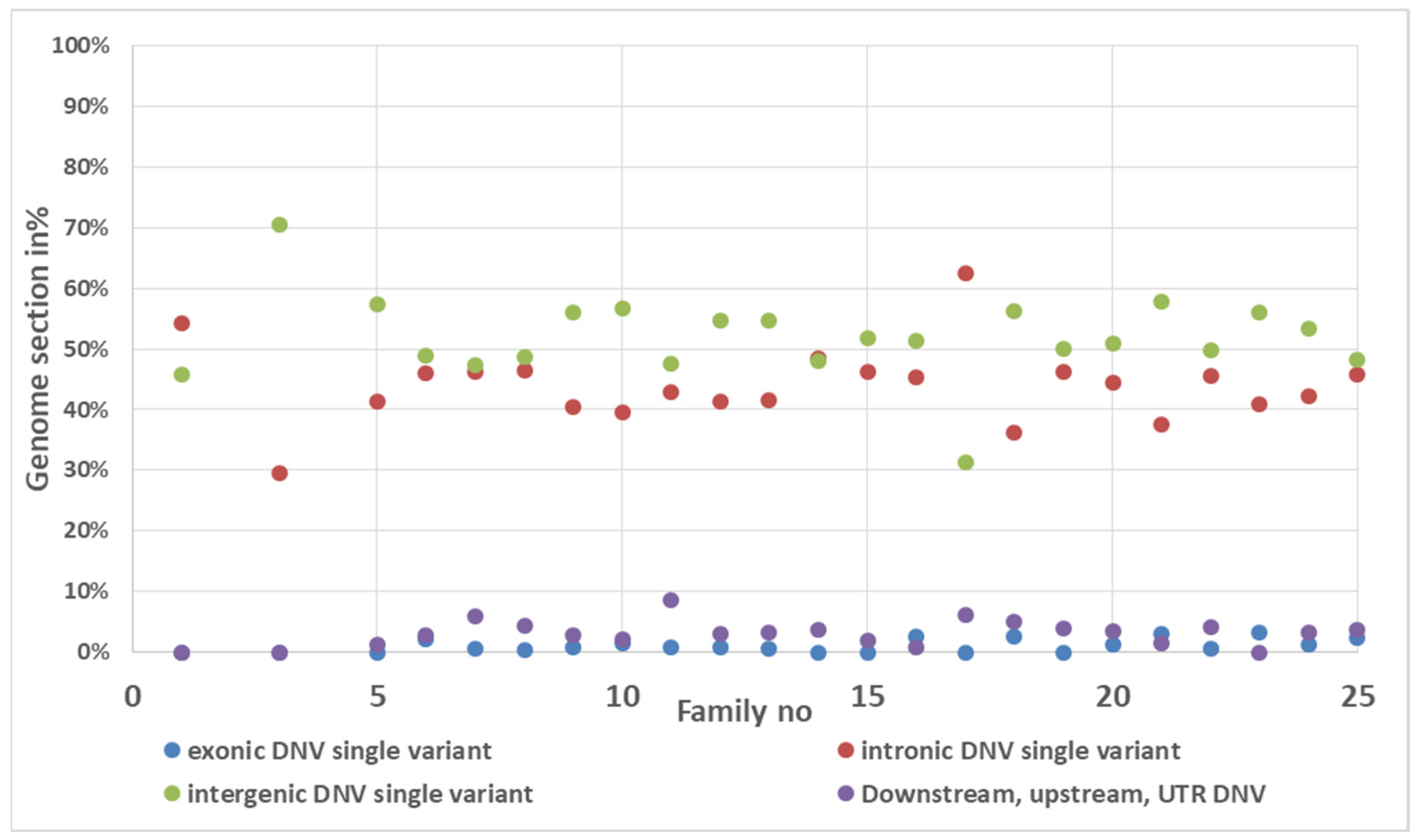
3.4. De Novo Mutation Discovery
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urnikyte, A.; Flores-Bello, A.; Mondal, M.; Molyte, A.; Comas, D.; Calafell, F.; Bosch, E.; Kučinskas, V. Patterns of Genetic Structure and Adaptive Positive Selection in the Lithuanian Population from High-Density SNP Data. Sci. Rep. 2019, 9, 9163. [Google Scholar] [CrossRef] [PubMed]
- Urnikyte, A.; Molyte, A.; Kučinskas, V. Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania. Genes 2021, 12, 1730. [Google Scholar] [CrossRef] [PubMed]
- Pranckėnienė, L.; Jakaitienė, A.; Ambrozaitytė, L.; Kavaliauskienė, I.; Kučinskas, V. Insights Into. Front. Genet. 2018, 9, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Howie, B.; McCarthy, S.; Memari, Y.; Walter, K.; Min, J.L.; Danecek, P.; Malerba, G.; Trabetti, E.; Zheng, H.F.; et al. Improved Imputation of Low-Frequency and Rare Variants Using the UK10K Haplotype Reference Panel. Nat. Commun. 2015, 6, 8111. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Bonham, V.L.; Brody, L.C.; Ginoza, M.E.C.; Hutter, C.M.; Manolio, T.A.; Green, E.D. Prioritizing Diversity in Human Genomics Research. Nat. Rev. Genet. 2018, 19, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Sinsheimer, J.S.; Novembre, J. Characterizing Bias in Population Genetic Inferences from Low-Coverage Sequencing Data. Mol. Biol. Evol. 2014, 31, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Pranckėnienė, L.; Kučinskas, V. The Relative Fitness of the de Novo Variants in General Lithuanian Population vs. in Individuals with Intellectual Disability. Eur. J. Hum. Genet. 2021, 30, 332–338. [Google Scholar] [CrossRef]
- Urnikyte, A.; Domarkiene, I.; Stoma, S.; Ambrozaityte, L.; Uktveryte, I.; Meskiene, R.; Kasiulevičius, V.; Burokiene, N.; Kučinskas, V. CNV Analysis in the Lithuanian Population. BMC Genet. 2016, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Urnikytė, A.; Molytė, A.; Kučinskas, V. Recent Effective Population Size Estimated from Segments of Identity by Descent in the Lithuanian Population. Anthropol. Sci. 2017, 125, 53–58. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A Fast and Accurate Adapter Trimmer for next-Generation Sequencing Paired-End Reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2022).
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid Detection of Structural Variants and Indels for Germline and Cancer Sequencing Applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen-the Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A Curated Collection of Structural Variation in the Human Genome. Nucleic Acids Res. 2013, 42, D986–D992. [Google Scholar] [CrossRef] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An Integrated Map of Structural Variation in 2,504 Human Genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and Characterization of Structural Variation in 17,795 Human Genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Children’s Mercy Research Institute Data. Available online: https://grch38.warehouse.cmh.edu/ (accessed on 1 March 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Project Data Processing the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly Austin 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. DbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An Accessible System for Testing SNV Novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT Missense Predictions for Genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Ranking Non-Synonymous Single Nucleotide Polymorphisms Based on Disease Concepts. Hum. Genom. 2014, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to Be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Sepal, A. Detection of Nonneutral Substitution Rates on Mammalian Phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying Novel Constrained Elements by Exploiting Biased Substitution Patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. DbSNP-Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information from over 60 000 Exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Li, K.; Li, B.; Wang, Z.; Fang, Z.; Wang, X.; Zhang, Y.; Luo, T.; Zhou, Q.; Wang, L.; et al. Gene4Denovo: An Integrated Database and Analytic Platform for de Novo Mutations in Humans. Nucleic Acids Res. 2019, 48, D913–D926. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. The Human Genome Project: Past, Present, and Future. Science 1990, 248, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.M.; Wasserman, W.W. The Genome Atlas: Navigating a New Era of Reference Genomes. Trends Genet. 2021, 37, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is It Time to Change the Reference Genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lee, W.P.; Ye, K.; Lee, C. One Reference Genome Is Not Enough. Genome Biol. 2019, 20, 104. [Google Scholar] [CrossRef] [Green Version]
- Zahn, L.M. The Human Genome. Science 2021, 373, 1458–1459. [Google Scholar] [CrossRef]
- Grytten, I.; Rand, K.D.; Nederbragt, A.J.; Sandve, G.K. Assessing Graph-Based Read Mappers against a Baseline Approach Highlights Strengths and Weaknesses of Current Methods. BMC Genom. 2020, 21, 282. [Google Scholar] [CrossRef]
- Glanzmann, B.; Jooste, T.; Ghoor, S.; Gordon, R.; Mia, R.; Mao, J.; Li, H.; Charls, P.; Douman, C.; Kotze, M.J.; et al. Human Whole Genome Sequencing in South Africa. Sci. Rep. 2021, 11, 606. [Google Scholar] [CrossRef]
- Peck, J.R.; Waxman, D. What Is Adaptation and How Should It Be Measured? J. Theor. Biol. 2018, 447, 190–198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Parents (n = 50) | Newborns (n = 25) | |
|---|---|---|
| Raw reads in M | 78,447 | 83,755 |
| Bases (Gb) | 11,832 | 12,633 |
| Coverage depth | 35.46 | 37.88 |
| SNVs | 4,704,096 | 4,696,226 |
| SNPs | 3,791,674 | 3,783,578 |
| Insertions (Hom) | 155,784 | 153,755 |
| Insertions (Het) | 277,288 | 276,262 |
| Deletions (Hom) | 152,914 | 150,766 |
| Deletions (Het) | 293,193 | 291,467 |
| Indels (Het) | 22,332 | 22,377 |
| Parents (n = 49) | Newborns (n = 24) | ||
|---|---|---|---|
| Statistics | Deletions | p-Value | |
| Mean | 4159.39 | 4191.33 | 0.5611 |
| SE | 33.63 | 47.08 | |
| Median | 4126.0 | 4172.5 | |
| Mode | 3963 | #N/A | |
| SD | 23,543 | 23,064 | |
| Duplications | |||
| Mean | 34,884 | 35,408 | 0.2747 |
| SE | 3.94 | 4.76 | |
| Median | 346 | 356 | |
| Mode | 365 | 355 | |
| SD | 27.61 | 23.31 | |
| Insertions | |||
| Mean | 4620.49 | 4661.54 | 0.5033 |
| SE | 49.98 | 63.05 | |
| Median | 4612.0 | 4697.5 | |
| Mode | #N/A | #N/A | |
| SD | 34,983 | 30,889 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urnikyte, A.; Pranckeniene, L.; Domarkiene, I.; Dauengauer-Kirliene, S.; Molyte, A.; Matuleviciene, A.; Pilypiene, I.; Kučinskas, V. Inherited and De Novo Variation in Lithuanian Genomes: Introduction to the Analysis of the Generational Shift. Genes 2022, 13, 569. https://doi.org/10.3390/genes13040569
Urnikyte A, Pranckeniene L, Domarkiene I, Dauengauer-Kirliene S, Molyte A, Matuleviciene A, Pilypiene I, Kučinskas V. Inherited and De Novo Variation in Lithuanian Genomes: Introduction to the Analysis of the Generational Shift. Genes. 2022; 13(4):569. https://doi.org/10.3390/genes13040569
Chicago/Turabian StyleUrnikyte, Alina, Laura Pranckeniene, Ingrida Domarkiene, Svetlana Dauengauer-Kirliene, Alma Molyte, Ausra Matuleviciene, Ingrida Pilypiene, and Vaidutis Kučinskas. 2022. "Inherited and De Novo Variation in Lithuanian Genomes: Introduction to the Analysis of the Generational Shift" Genes 13, no. 4: 569. https://doi.org/10.3390/genes13040569
APA StyleUrnikyte, A., Pranckeniene, L., Domarkiene, I., Dauengauer-Kirliene, S., Molyte, A., Matuleviciene, A., Pilypiene, I., & Kučinskas, V. (2022). Inherited and De Novo Variation in Lithuanian Genomes: Introduction to the Analysis of the Generational Shift. Genes, 13(4), 569. https://doi.org/10.3390/genes13040569