
Identification of a Cancer-Predisposing Germline POT1 p.Ile49Metfs*7 Variant by Targeted Sequencing of a Splenic Marginal Zone Lymphoma

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lymphoid NGS of Bone Marrow
2.2. NGS of Skin Fibroblasts for Confirmatory Germline Testing
2.3. Fluorescence In Situ Hybridization (FISH) Analysis of Bone Marrow Aspirate
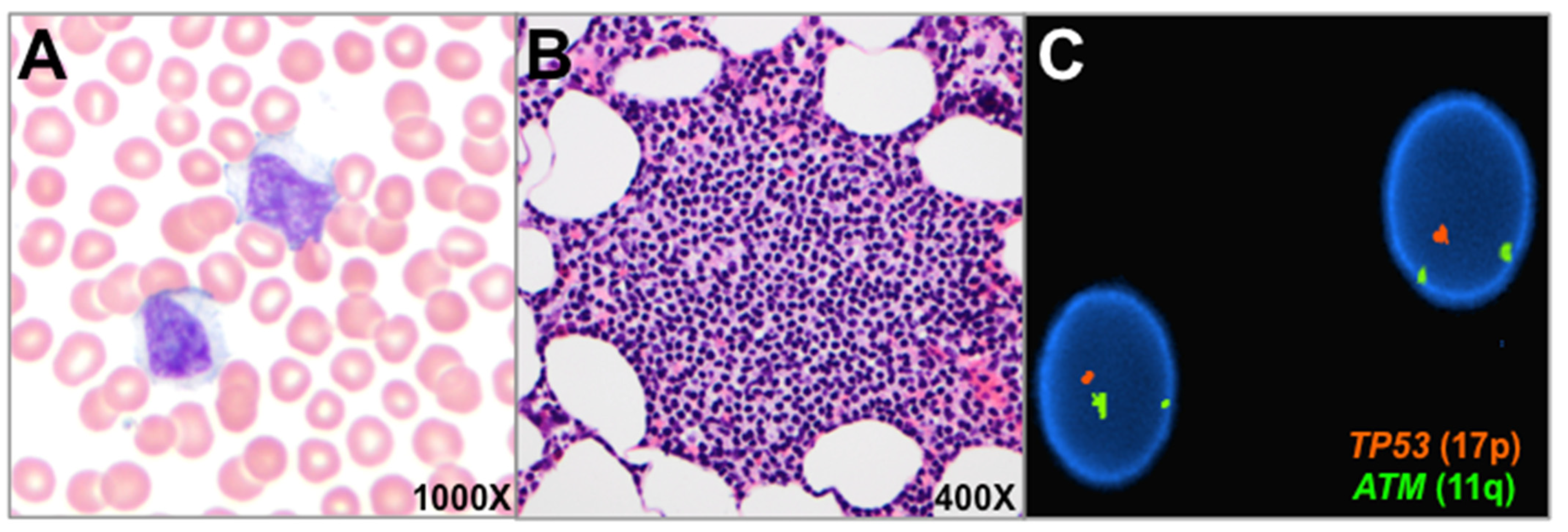
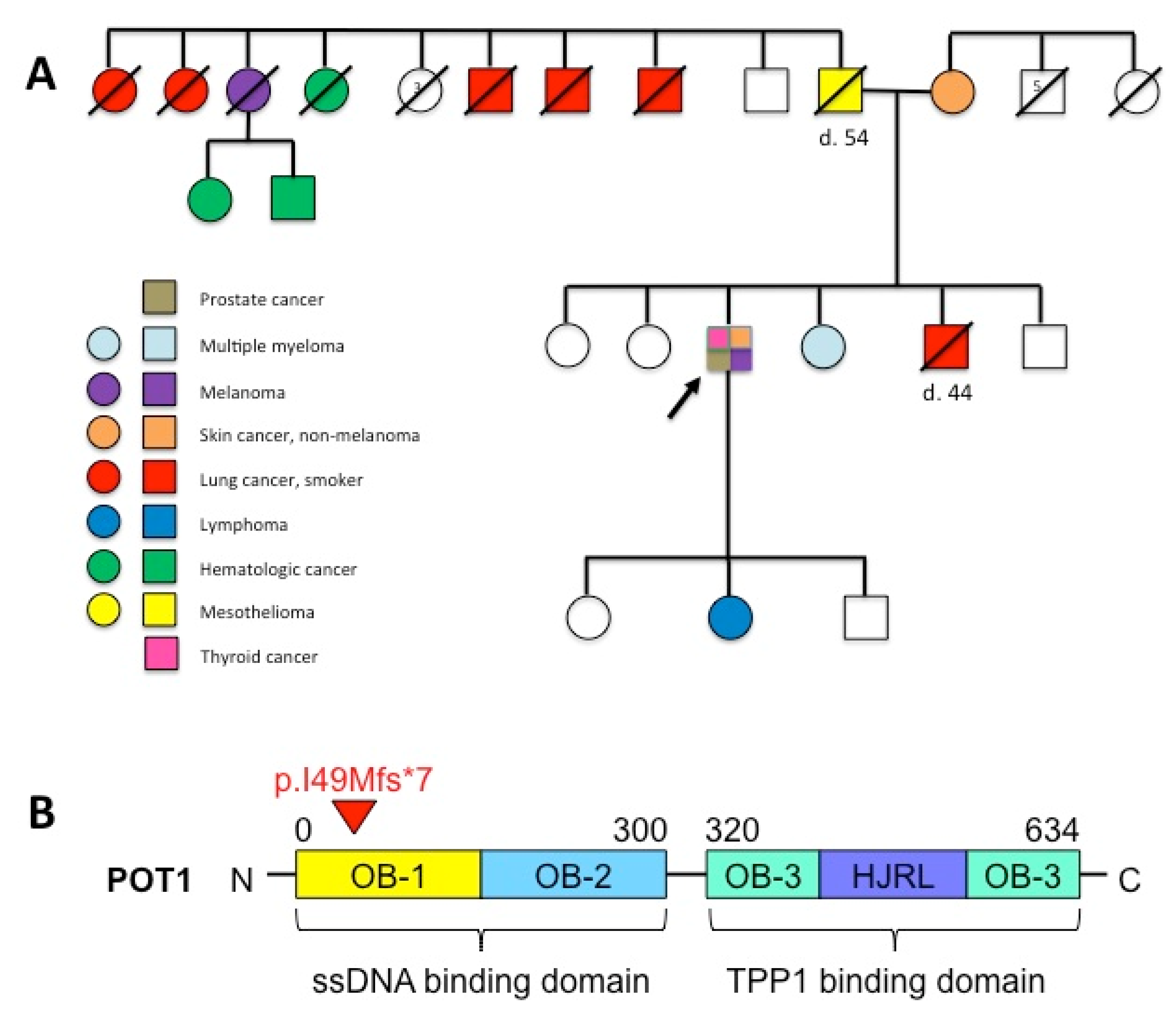
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martínez-Trillos, A.; Villamor, N.; Rodríguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Speedy, H.E.; Kinnersley, B.; Chubb, D.; Broderick, P.; Law, P.J.; Litchfield, K.; Jayne, S.; Dyer, M.J.S.; Dearden, C.; Follows, G.A.; et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016, 128, 2319–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljaket, M.; et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMaster, M.L.; Sun, C.; Landi, M.T.; Savage, S.A.; Rotunno, M.; Yang, X.R.; Jones, K.; Vogt, A.; Hutchinson, A.; Zhu, B.; et al. Germline mutations in protection of telomeres 1 in two families with hodgkin lymphoma. Br. J. Haematol. 2018, 181, 372–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Calvete, O.; Martinez, P.; Garcia-Pavia, P.; Benitez-Buelga, C.; Paumard-Hernández, B.; Fernandez, V.; Dominguez, F.; Salas, C.; Romero-Laorden, N.; Garcia-Donas, J.; et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative li-fraumeni-like families. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, B.H.; Getta, B.; Dietrich, S.; Taylor, J.; Won, H.; Bogenberger, J.M.; Scott, S.; Kim, E.; Chung, Y.R.; Chung, S.S.; et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017, 130, 1644–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterfall, J.J.; Arons, E.; Walker, R.L.; Pineda, M.; Roth, L.; Killian, J.K.; Abaan, O.D.; Davis, S.R.; Kreitman, R.J.; Meltzer, P.S. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat. Genet. 2014, 46, 8–10. [Google Scholar] [CrossRef]
- Spina, V.; Khiabanian, H.; Messina, M.; Monti, S.; Cascione, L.; Bruscaggin, A.; Spaccarotella, E.; Holmes, A.B.; Arcaini, L.; Lucioni, M.; et al. The genetics of nodal marginal zone lymphoma. Blood 2016, 128, 1362–1373. [Google Scholar] [CrossRef]
- Jajosky, A.N.; Havens, N.; Sadri, N.; Oduro, K.A.; Moore, E.M.; Beck, R.C.; Meyerson, H.J. Clinical utility of targeted next generation sequencing in the evaluation of low-grade lymphoproliferative disorders arising from the blood and bone marrow. Am. J. Clin. Pathol. 2021, 156, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Hennick, K.; Johnson, J.; Finnerty, B.; Choo, S.; Short, S.B.; Drubin, C.; Forster, R.; McMaster, M.L.; Hockemeyer, D. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021, 40, e107346. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Lakamp-Hawley, A.S.; Kolar, C.; Yan, Y.; Borgstahl, G.E.O.; Ouellette, M.M. The OB-fold domain 1 of human POT1 recognizes both telomeric and non-telomeric DNA motifs. Biochimie 2015, 115, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, A.; Mian, M.; Chigrinova, E.; Arcaini, L.; Bhagat, G.; Novak, U.; Rancoita, P.M.V.; De Campos, C.P.; Forconi, F.; Gascoyne, R.D.; et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood 2011, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Nathan, V.; Johansson, P.A.; Palmer, J.M.; Hamilton, H.R.; Howlie, M.; Brooks, K.M.; Hayward, N.K.; Pritchard, A.L. A rare missense variant in protection of telomeres 1 (POT1) predisposes to a range of haematological malignancies. Br. J. Haematol. 2021, 192, e57–e60. [Google Scholar] [CrossRef]
- Lim, T.L.; Lieberman, D.B.; Davis, A.R.; Loren, A.W.; Hausler, R.; Bigdeli, A.; Li, Y.; Powers, J.; Raper, A.; Center, R.G.; et al. Germline POT1 variants can predispose to myeloid and lymphoid neoplasms. Leukemia 2022, 36, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Michler, P.; Schedel, A.; Witschas, M.; Friedrich, U.A.; Wagener, R.; Mehtonen, J.; Brozou, T.; Menzel, M.; Walter, C.; Nabi, D.; et al. Germline POT1 deregulation can predispose to myeloid malignancies in childhood. Int. J. Mol. Sci. 2021, 22, 11572. [Google Scholar] [CrossRef] [PubMed]
- Salido, M.; Baro, C.; Oscier, D.; Stamatopoulos, K.; Dierlamm, J.; Matutes, E.; Traverse-Glehen, A.; Berger, F.; Felman, P.; Thieblemont, C.; et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: A multicenter study of the Splenic B-Cell Lymphoma Group. Blood 2010, 116, 1479–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, F.; Cho-Vega, J.H.; Lennon, P.A.; Madan, G.; Luthra, J.; Bailey, M.; Breeden, D.; Jones, L.; Medeiros, J.; Luthra, R. Splenic marginal zone lymphomas are characterized by loss of interstitial regions of chromosome 7q, 7q31.32 and 7q36.2 that include the protection of telomere 1 (POT1) and sonic hedgehog (SHH) genes. Br. J. Haematol. 2008, 142, 216–226. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variant | VAF | Tissue Tested | Origin | COSMIC Database Frequency | ClinVar ID, gnomAD Population Frequency | Pathogenicity (ACMG Criteria) |
|---|---|---|---|---|---|---|
| TRAF3 c.501_502delGA, p.Lys168Glyfs*3 | 8% | bone marrow | presumed somatic | absent | absent from ClinVar and gnomAD | likely pathogenic |
| POT1 c.147delT, p.Ile49Metfs*7 | 51% | bone marrow and skin fibroblasts | confirmed germline | absent | ClinVar Variation ID: 420174; 0.007% (gnomAD) | likely pathogenic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jajosky, A.N.; Mitchell, A.L.; Akgul, M.; Shetty, S.; Yoest, J.M.; Gerson, S.L.; Sadri, N.; Oduro, K.A., Jr. Identification of a Cancer-Predisposing Germline POT1 p.Ile49Metfs*7 Variant by Targeted Sequencing of a Splenic Marginal Zone Lymphoma. Genes 2022, 13, 591. https://doi.org/10.3390/genes13040591
Jajosky AN, Mitchell AL, Akgul M, Shetty S, Yoest JM, Gerson SL, Sadri N, Oduro KA Jr. Identification of a Cancer-Predisposing Germline POT1 p.Ile49Metfs*7 Variant by Targeted Sequencing of a Splenic Marginal Zone Lymphoma. Genes. 2022; 13(4):591. https://doi.org/10.3390/genes13040591
Chicago/Turabian StyleJajosky, Audrey N., Anna L. Mitchell, Mahmut Akgul, Shashirekha Shetty, Jennifer M. Yoest, Stanton L. Gerson, Navid Sadri, and Kwadwo A. Oduro, Jr. 2022. "Identification of a Cancer-Predisposing Germline POT1 p.Ile49Metfs*7 Variant by Targeted Sequencing of a Splenic Marginal Zone Lymphoma" Genes 13, no. 4: 591. https://doi.org/10.3390/genes13040591
APA StyleJajosky, A. N., Mitchell, A. L., Akgul, M., Shetty, S., Yoest, J. M., Gerson, S. L., Sadri, N., & Oduro, K. A., Jr. (2022). Identification of a Cancer-Predisposing Germline POT1 p.Ile49Metfs*7 Variant by Targeted Sequencing of a Splenic Marginal Zone Lymphoma. Genes, 13(4), 591. https://doi.org/10.3390/genes13040591