
Pediatric High Grade Glioma Classification Criteria and Molecular Features of a Case Series

 , ,
, ,
Abstract
:1. Introduction
1.1. Histone H3 Mutant pHGGs
1.2. IDH Mutant pHGGs
1.3. H3/IDH Wild Type pHGGS
1.4. PXA-like pHGGs
1.5. pHGGs Amplified in NMYC, PDGFRA or EGFR
1.6. Hyper Mutant pHGGs
1.7. Infant-Type Hemispheric Gliomas
1.8. Diffuse Midline Gliomas EGFR Mutant
1.9. Aim
2. Patients and Methods
2.1. Patients
2.2. Methods
2.3. DNA Extraction
2.4. Gene Panel and Bioinformatics Analysis of pHGG Panel Genes
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. 10), x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Pfister, S.M.; Jones, D.T.W. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. 3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Thorbinson, C.; Kilday, J.P. Childhood Malignant Brain Tumors: Balancing the Bench and Bedside. Cancers 2021, 13, 6099. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef]
- Kim, Y.Z. Altered histone modifications in gliomas. Brain Tumor Res. Treat. 2014, 2, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillac, C.; Thomas, C.; Dastagirzada, Y.; Hidalgo, E.T.; Golfinos, J.G.; Zagzag, D.; Wisoff, J.H.; Karajannis, M.A.; Snuderl, M. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol. Commun. 2016, 4, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, G.; Oberheim Bush, N.A.; Berger, M.S.; Perry, A.; Solomon, D.A. Diffuse non-midline glioma with H3F3A K27M mutation: A prognostic and treatment dilemma. Acta Neuropathol. Commun. 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Toenjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.Y.; Won, J.K.; Park, C.K.; Kim, S.K.; Choi, S.H.; Kim, T.; Yun, H.; Park, S.H. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2021, 38, 4–13. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Buccoliero, A.M.; Castiglione, F.; Degl’Innocenti, D.R.; Gheri, C.F.; Genitori, L.; Taddei, G.L. IDH1 mutation in pediatric gliomas: Has it a diagnostic and prognostic value? Fetal. Pediatr. Pathol. 2012, 31, 278–282. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Sumerauer, D.; Krskova, L.; Vicha, A.; Misove, A.; Mamatjan, Y.; Jencova, P.; Vlckova, M.; Slamova, L.; Vanova, K.; Liby, P.; et al. Rare IDH1 variants are common in pediatric hemispheric diffuse astrocytomas and frequently associated with Li-Fraumeni syndrome. Acta Neuropathol. 2020, 139, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Tauziède-Espariat, A.; Debily, M.A.; Castel, D.; Grill, J.; Puget, S.; Sabel, M.; Blomgren, K.; Gareton, A.; Dangouloff-Ros, V.; Lechapt, E.; et al. An integrative radiological, histopathological and molecular analysis of pediatric pontine histone-wildtype glioma with MYCN amplification (HGG-MYCN). Acta Neuropathol. Commun. 2019, 7, 87. [Google Scholar] [CrossRef]
- Koschmann, C.; Zamler, D.; MacKay, A.; Robinson, D.; Wu, Y.M.; Doherty, R.; Marini, B.; Tran, D.; Garton, H.; Muraszko, K.; et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 2016, 4, 65696–65706. [Google Scholar] [CrossRef] [Green Version]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Viana-Pereira, M.; Lee, A.; Popov, S.; Bax, D.A.; Al-Sarraj, S.; Bridges, L.R.; Stávale, J.N.; Hargrave, D.; Jones, C.; Reis, R.M. Microsatellite instability in pediatric high grade glioma is associated with genomic profile and differential target gene inactivation. PLoS ONE 2011, 6, e20588. [Google Scholar] [CrossRef] [Green Version]
- Alphones, S.; Chatterjee, U.; Singh, A.; Das, A.; Zameer, L.; Achari, R.; Bhattacharya, A.; Roy, P. Immunohistochemical screening for mismatch repair protein deficiency in paediatric high-grade gliomas-institutional experience and review of literature. Childs Nerv. Syst. 2021, 37, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.A.; Stechishin, O.D.; Luchman, H.A.; Lun, X.Q.; Senger, D.L.; Robbins, S.M.; Cairncross, J.G.; Weiss, S. Novel MSH6 mutations in treatment-naïve glioblastoma and anaplastic oligodendroglioma contribute to temozolomide resistance independently of MGMT promoter methylation. Clin. Cancer Res. 2014, 20, 4894–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quanye, S.; Chunying, P.; Qiuyuan, L.; Tianxiu, D.; Yucui, D.; Wenjing, X.; Peng, Z.; Yujiao, G.; Ziqi, Z.; Yifan, G.; et al. Up-regulation of MSH6 is associated with temozolomide resistance in human glioblastoma. Biochem. Biophys. Res. Commun. 2018, 496, 1040–1046. [Google Scholar]
- Yamashiro, K.; Nakao, K.; Ohba, S.; Hirose, Y. Human Glioma Cells Acquire Temozolomide Resistance After Repeated Drug Exposure Via DNA Mismatch Repair Dysfunction. Anticancer Res. 2020, 40, 1315–1323. [Google Scholar] [CrossRef]
- Sievers, P.; Sill, M.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Sturm, D.; Hench, J.; Frank, S.; Krskova, L.; Vicha, A.; et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. 2021, 23, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020, 39, 1109–1113. [Google Scholar] [CrossRef]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735.e7. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. Lyon (France): International Agency for Research on Cancer, 5th ed.; WHO Classification of Tumours Series; WHO: Geneva, Switzerland, 2021; Volume 6. [Google Scholar]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point variants in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.Z.; Jennings, L.J.; Wadhwani, N.R.; Brat, D.J.; Horbinski, C.M. The Essentials of Molecular Testing in CNS Tumors: What to Order and How to Integrate Results. Curr. Neurol. Neurosci. Rep. 2020, 20, 23. [Google Scholar] [CrossRef]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J. Neurooncol. 2017, 134, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Bready, D.; Placantonakis, D.G. Molecular Pathogenesis of Low-Grade Glioma. Neurosurg. Clin. N. Am. 2019, 30, 17–25. [Google Scholar] [CrossRef]
- Suri, V.; Das, P.; Pathak, P.; Jain, A.; Sharma, M.C.; Borkar, S.A.; Suri, A.; Gupta, D.; Sarkar, C. Pediatric glioblastomas: A histopathological and molecular genetic study. Neuro Oncol. 2009, 11, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallappagoudar, S.; Yadav, R.K.; Lowe, B.R.; Partridge, J.F. Histone H3 mutations—A special role for H3.3 in tumorigenesis? Chromosoma 2015, 124, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.; Hoeman, C.; Becher, O. Children are not just little adults: Recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014, 75, 205–209. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Kobayashi, S.; Costa, D.B. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Nafa, K.; Chaft, J.E.; Rekhtman, N.; Lau, C.; Reva, B.A.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. EGFR exon 20 insertion mutations in lung adenocarcinomas: Prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol. Cancer Ther. 2013, 12, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Akhavanfard, S.; Yehia, L.; Padmanabhan, R.; Reynolds, J.P.; Ni, Y.; Eng, C. Germline EGFR variants are over-represented in adolescents and young adults (AYA) with adrenocortical carcinoma. Hum. Mol. Genet. 2021, 29, 3679–3690. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hübner, J.M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Ellison Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018, 13, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Hübner, J.M.; Müller, T.; Papageorgiou, D.N.; Mauermann, M.; Krijgsveld, J.; Russell, R.B.; Ellison, D.W.; Pfister, S.M.; Pajtler, K.W.; Kool, M. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019, 21, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Y.; Liu, X.; Li, V.K.; Ding, Y.; Yang, B.; Geng, J.; Lai, R.; Ding, S.; Ni, M.; Zhao, R. Detection of mismatch repair gene germline mutation carrier among Chinese population with colorectal cancer. BMC Cancer 2008, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumal Kumar, D.; Sneha, P.; Uppin, J.; Usha, S.; George Priya Doss, C. Investigating the Influence of Hotspot Mutations in Protein-Protein Interaction of IDH1 Homodimer Protein: A Computational Approach. Adv. Protein Chem. Struct. Biol. 2018, 111, 243–261. [Google Scholar]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [Green Version]
- Loo, E.; Khalili, P.; Beuhler, K.; Siddiqi, I.; Vasef, M.A. BRAF V600E Mutation Across Multiple Tumor Types: Correlation Between DNA-based Sequencing and Mutation-specific Immunohistochemistry. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 709–713. [Google Scholar] [CrossRef]
- Raynaud, S.; Carbuccia, N.; Colin, C.; Adélaïde, J.; Mozziconacci, M.J.; Metellus, P.; Chinot, O.; Birnbaum, D.; Chaffanet, M.; Figarella-Branger, D. Absence of R140Q mutation of isocitrate dehydrogenase 2 in gliomas and breast cancers. Oncol. Lett. 2010, 1, 883–884. [Google Scholar] [CrossRef]
- Nambirajan, A.; Sharma, A.; Rajeshwari, M.; Boorgula, M.T.; Doddamani, R.; Garg, A.; Suri, V.; Sarkar, C.; Sharma, M.C. EZH2 inhibitory protein (EZHIP/Cxorf67) expression correlates strongly with H3K27me3 loss in posterior fossa ependymomas and is mutually exclusive with H3K27M mutations. Brain Tumor Pathol. 2021, 38, 30–40. [Google Scholar] [CrossRef]



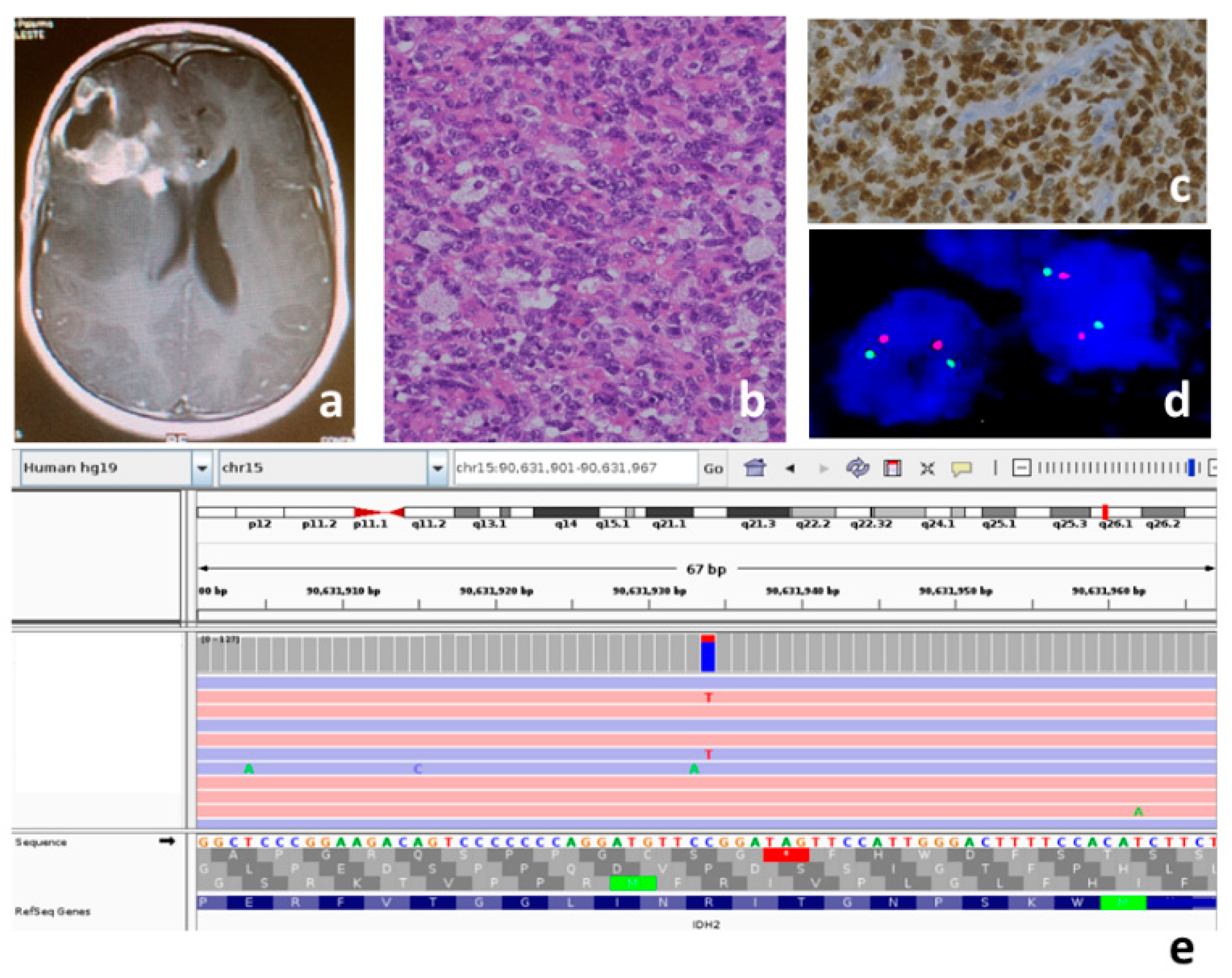{kind=link}
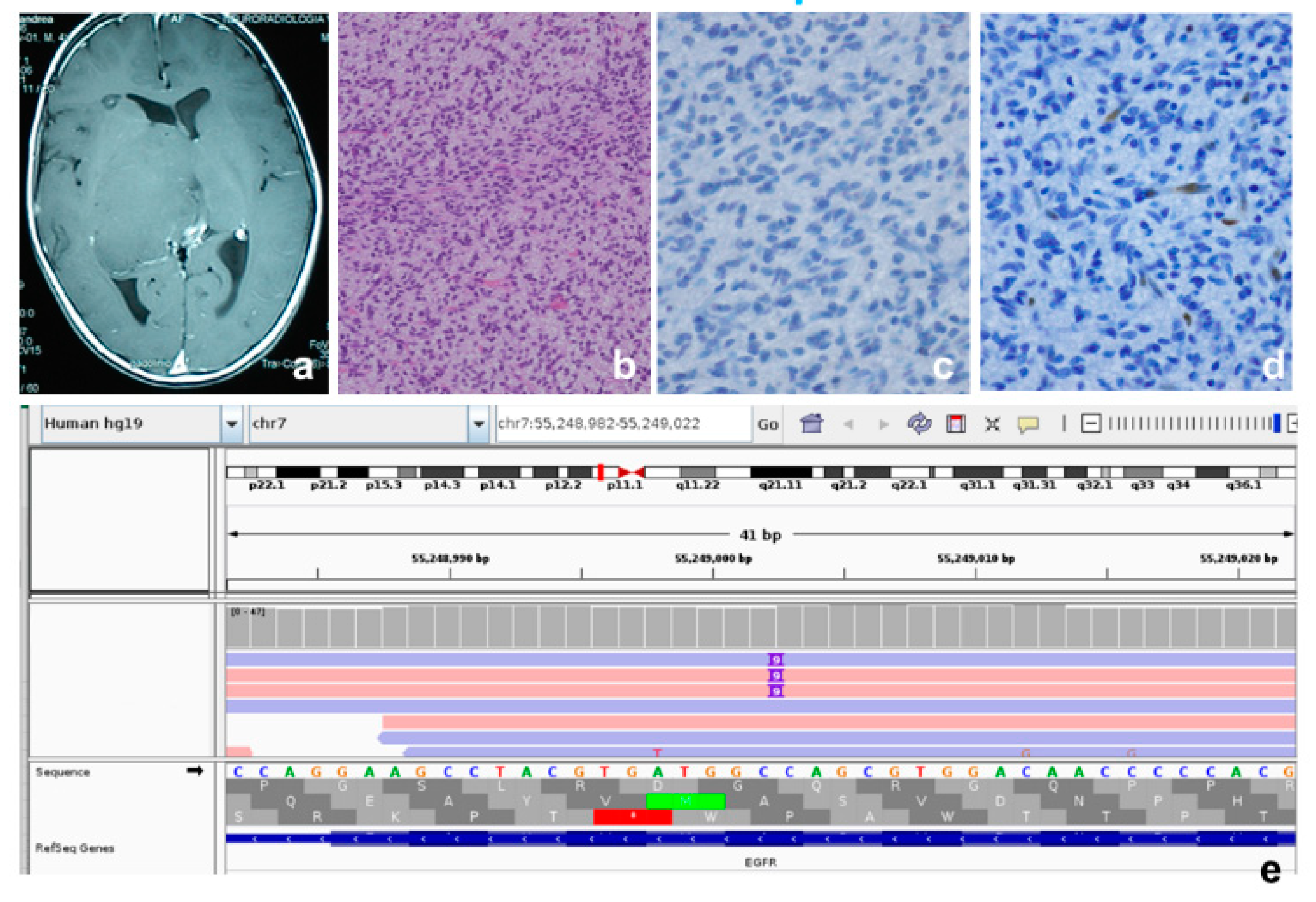{kind=link}
| Case | Age (Years) | Sex | Localization | Molecular Classification |
|---|---|---|---|---|
| P1 | 0 | F | Hemispheric | Not determined |
| P2 | 9 | M | Thalamus | H3.3K27 mutant |
| P3 | 12 | F | Hemispheric | IDH mutant |
| P4 | 4 | M | Thalamus | EGFR mutant |
| P5 | 8 | F | C3–C4 | H3.3K27 mutant |
| P6 | 5 | M | Hypothamalus | PXA-like |
| P7 | 10 | F | Hemispheric | Not determined |
| P8 | 8 | F | Cerebellum | Not determined |
| P9 | 13 | M | Midbrain | H3.3K27 mutant |
| P10 | 18 | M | Hemispheric | H3.3G34 mutant |
| P11 | 14 | F | Hemispheric | H3.3G34 mutant |
| Case | Genes | Transcript | cDNA | Protein | Exon | Allele Frequency | Variant Coverage | ACMG Classification | COSMIC |
|---|---|---|---|---|---|---|---|---|---|
| P1 | EZHIP | NM_203407 | c.1328C>T | p.Ser443Phe | 1 | 1.09 × 10−5 | 119X | Uncertain Significance | - |
| P2 | H3F3A | NM_002107 | c.83A>T | p.Lys27Met | 2 | - | 150X | Pathogenic | COSM327928 |
| TP53 | NM_000546 | c.537T>G | p.His179Gln | 5 | - | 41X | Pathogenic | COSM11249 | |
| TP53 | NM_000546 | c.329G>T | p.Arg110Leu | 4 | - | 44X | Pathogenic | COSM10716 | |
| P3 | H3F3A | NM_002107 | c.246T>G | p.Asp82Glu | 3 | - | 90X | Uncertain Significance LP | - |
| HRAS | NM_005343 | c.287A>G | p.Tyr96Cys | 3 | - | 57X | Uncertain Significance | - | |
| HRAS | NM_005343 | c.413G>A | p.Gly138Asp | 4 | - | 83X | Uncertain Significance | - | |
| IDH2 | NM_002168 | c.419G>A | p.Arg140Gln | 4 | - | 119X | Pathogenic | COSM41590 | |
| PDGFRA | NM_006206 | c.1973T>C | p.Val658Ala | 14 | - | 67X | Likely Pathogenic | - | |
| PDGFRA | NM_006206 | c.236G>A | p.Gly79Asp | 3 | 8.91 × 10−3 | 145X | Benign | COSM5019287 | |
| TERT | NM_198253 | c.863C>T | p.Ala288Val | 2 | 4.86 × 10−5 | 53X | Uncertain Significance | - | |
| TP53 | NM_000546 | c.541C>T | p.Arg181Cys | 5 | - | 24X | Pathogenic | COSM11090 | |
| TP53 | NM_000546 | c.916C>T | p.Arg306 * | 8 | - | 97X | Pathogenic | COSM10663 | |
| P4 | EGFR | NM_005228 | c.2300_2301ins9 | p.Ala767_Ser768ins3 | 20 | - | 42X | Pathogenic | - |
| P5 | H3F3A | NM_002107 | c.83A>T | p.Lys27Met | 2 | - | 119X | Pathogenic | COSM327928 |
| NF1 | NM_000267 | c.5242C>T | p.Arg1748 * | 37 | - | 83X | Pathogenic | - | |
| P6 | BRAF | NM_004333 | c.1799T>A | p.Val600Glu | 15 | 3.98 × 10−6 | 171X | Pathogenic | COSM476 |
| PDGFRA | NM_006206 | c.1285G>A | p.Gly429Arg | 9 | 3.43 × 10−4 | 55X | Uncertain Significance | - | |
| P7 | EGFR | NM_005228 | c.1994G>A | p.Gly665Asp | 17 | - | 53X | Uncertain Significance | COSM8256230 |
| EGFR | NM_005228 | c.1973T>A | p.Leu658Gln | 17 | - | 64X | Uncertain Significance LP | COSM6354678 | |
| P8 | TP53 | NM_000546 | c.1047_1048ins13 | p.Leu350_K351ins * | 10 | - | 52X | Pathogenic | - |
| TP53 | NM_000546 | c.451C>A | p.Pro151Thr | 5 | - | 41X | Pathogenic | COSM43911 | |
| P9 | H3F3A | NM_002107 | c.83A>T | p.Lys27Met | 2 | - | 155X | Pathogenic | COSM327928 |
| IDH1 | NM_005896 | c.532G>A | p.Val178Ile | 6 | 4.95 × 10−2 | 111X | Benign | COSM97131 | |
| TP53 | NM_000546 | c.817C>T | p.Arg273Cys | 8 | 1.20 × 10−5 | 44X | Pathogenic | COSM10659 | |
| P10 | H3F3A | NM_002107 | c.103G>A | p.Gly34Arg | 2 | - | 100X | Pathogenic | COSM327929 |
| MSH2 | NM_000251 | c.1145G>A | p.Arg382His | 7 | 7.95 × 10−6 | 97X | Uncertain Significance LP | COSM3185920 | |
| PTEN | NM_000314 | c.302delT | p.Iso101fs (LOH) | 5 | - | 69X | Pathogenic | - | |
| TP53 | NM_000546 | c.574C>T | p.Gln192 * (LOH) | 6 | - | 79X | Pathogenic | COSM10733 | |
| P11 | ATRX | NM_00489 | c.2341C>T | p.Arg781 * | 9 | - | 130X | Pathogenic | COSM1716656 |
| EGFR | NM_005228 | c.2024G>A | p.Arg675Gln | 17 | 2.11 × 10−4 | 540X | Uncertain Significance | COSM6938266 | |
| H3F3A | NM_002107 | c.104G>T | p.Gly34Val | 2 | - | 26X | Pathogenic | COSM502595 | |
| PIK3CA | NM_006218 | c.1633G>A | p.Glu545Lys | 10 | - | 81X | Pathogenic | COSM763 | |
| PTEN | NM_000314 | c.781C>G | p.Gln261Glu (LOH) | 7 | - | 35X | Uncertain Significance LP | - | |
| TP53 | NM_000546 | c.637C>T | p.Arg213 * | 6 | - | 152X | Pathogenic | COSM10654 | |
| TP53 | NM_000546 | c.1024C>T | p.Arg342 * | 10 | - | 106X | Pathogenic | COSM11073 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buccoliero, A.M.; Giunti, L.; Moscardi, S.; Castiglione, F.; Provenzano, A.; Sardi, I.; Scagnet, M.; Genitori, L.; Caporalini, C. Pediatric High Grade Glioma Classification Criteria and Molecular Features of a Case Series. Genes 2022, 13, 624. https://doi.org/10.3390/genes13040624
Buccoliero AM, Giunti L, Moscardi S, Castiglione F, Provenzano A, Sardi I, Scagnet M, Genitori L, Caporalini C. Pediatric High Grade Glioma Classification Criteria and Molecular Features of a Case Series. Genes. 2022; 13(4):624. https://doi.org/10.3390/genes13040624
Chicago/Turabian StyleBuccoliero, Anna Maria, Laura Giunti, Selene Moscardi, Francesca Castiglione, Aldesia Provenzano, Iacopo Sardi, Mirko Scagnet, Lorenzo Genitori, and Chiara Caporalini. 2022. "Pediatric High Grade Glioma Classification Criteria and Molecular Features of a Case Series" Genes 13, no. 4: 624. https://doi.org/10.3390/genes13040624
APA StyleBuccoliero, A. M., Giunti, L., Moscardi, S., Castiglione, F., Provenzano, A., Sardi, I., Scagnet, M., Genitori, L., & Caporalini, C. (2022). Pediatric High Grade Glioma Classification Criteria and Molecular Features of a Case Series. Genes, 13(4), 624. https://doi.org/10.3390/genes13040624